



1. Wstępne zrozumienie
Na tym etapie musimy zrozumieć pewne pojęcia i terminologię, aby nie popełnić błędów w obecności naszych seniorów, takich jak:
P: Jaka jest różnica między RT-PCR, qPCR, Real-time PCR i RT-PCR w czasie rzeczywistym?
Odpowiedź: RT-PCR to PCR z odwrotną transkrypcją(PCR z odwrotną transkrypcją, RT-PCR), która jest szeroko stosowaną odmianą reakcji łańcuchowej polimerazy (PCR).W RT-PCR nić RNA jest poddawana odwrotnej transkrypcji do komplementarnego DNA, który jest następnie używany jako matryca do amplifikacji DNA metodą PCR.
PCR w czasie rzeczywistym i qPCR(Quantitative Rea-ltime-PCR) to to samo, oba są ilościowymi PCR w czasie rzeczywistym, co oznacza, że każdy cykl PCR ma rekordy danych w czasie rzeczywistym, więc liczbę początkowych szablonów można dostosować do dokładnej analizy.
Chociaż zarówno PCR w czasie rzeczywistym (ilościowy PCR fluorescencyjny w czasie rzeczywistym), jak i PCR z odwrotną transkrypcją (PCR z odwrotną transkrypcją) wydają się być określane skrótem RT-PCR, międzynarodowa konwencja jest następująca: RT-PCR odnosi się konkretnie do odwrotnej transkrypcjiPCR , PCR w czasie rzeczywistym jest ogólnie określany skrótem qPCR (ilościowy PCR w czasie rzeczywistym).
I RT-PCR w czasie rzeczywistym (RT-qPCR), jest to PCR z odwrotną transkrypcją w połączeniu z fluorescencyjną technologią ilościową: najpierw uzyskaj cDNA (RT) z odwrotnej transkrypcji RNA, a następnie użyj Real-time PCR do analizy ilościowej (qPCR).Większość laboratoriów wykonuje RT-qPCR, czyli badania nad obniżeniem ekspresji RNA, więc qPCR, o którym wszyscy mówią w laboratorium, w rzeczywistości odnosi się do RT-qPCR, ale nie zapominaj, że wciąż istnieje wiele testów DNA w zastosowaniach klinicznych.Analiza ilościowa, taka jak wykrywanie wirusa zapalenia wątroby typu B HBV.
Pytanie: Po przeczytaniu wielu fluorescencyjnych ilościowych PCR, dlaczego zamplifikowany fragment powinien być kontrolowany w zakresie 80-300 bp?
Odpowiedź: Długość każdej sekwencji genów jest inna, niektóre mają kilka kb, niektóre setki pz, ale potrzebujemy tylko wymagać, aby długość produktu wynosiła 80-300 pz podczas projektowania starterów, zbyt krótkie lub zbyt długie nie są odpowiednie do fluorescencyjnej ilościowej detekcji PCR.Fragment produktu jest zbyt krótki, aby można go było odróżnić od startera-dimeru.Długość startera-dimeru wynosi około 30-40 pz i trudno jest rozróżnić, czy jest to starter-dimer, czy produkt, jeśli jest mniejszy niż 80 pz.Jeśli fragment produktu jest zbyt długi, przekraczający 300 bp, łatwo doprowadzi to do niskiej wydajności amplifikacji i nie będzie w stanie skutecznie wykryć ilości genu.
Na przykład, kiedy liczysz, ile osób jest w klasie, wystarczy policzyć, ile jest ust.To samo dotyczy wykrywania genów, wystarczy wykryć określoną sekwencję genu, aby reprezentować całą sekwencję.Jeśli chcesz policzyć ludzi, musisz policzyć usta i nosy, uszy i okulary, a łatwo popełnić błąd.
Aby rozszerzyć, w badaniach biologicznych istnieje wiele przypadków badawczych z punktu do obszaru, ponieważ sekwencja genu dowolnego gatunku jest bardzo długa, nie jest konieczne i niemożliwe zmierzenie wszystkich fragmentów, jak np.
P: Jaka jest optymalna długość do projektowania starterów qPCR?
Odpowiedź: Ogólnie rzecz biorąc, długość startera wynosi około 20-24 bp, co jest lepsze.Oczywiście przy projektowaniu podkładu musimy zwrócić uwagę na wartość TM podkładu, ponieważ jest to związane z optymalną temperaturą wyżarzania.Po wielu eksperymentach udowodniono, że 60°C jest lepszą wartością TM.Jeśli temperatura hybrydyzacji jest zbyt niska, łatwo doprowadzi to do niespecyficznej amplifikacji.Jeśli temperatura wyżarzania jest zbyt wysoka, wydajność amplifikacji będzie stosunkowo niska, szczyt krzywej amplifikacji rozpocznie się później, a wartość CT będzie opóźniona.
P: Czym różni się metoda barwienia od metody sondy?
Odpowiedź: metoda barwieniaNiektóre barwniki fluorescencyjne, takie jak SYBR Green Ⅰ, PicoGreen, BEBO itp., same nie emitują światła, ale emitują fluorescencję po związaniu się z mniejszym rowkiem dwuniciowego DNA.Dlatego na początku reakcji PCR maszyna nie może wykryć sygnału fluorescencyjnego.Kiedy reakcja osiąga etap wygrzewania-wydłużania, podwójna nić jest otwierana i nowa nić jest syntetyzowana pod działaniem polimerazy DNA, a cząsteczka fluorescencyjna wiąże się z mniejszym rowkiem dsDNA.Wraz ze wzrostem liczby cykli PCR coraz więcej barwników łączy się z dwuniciowym DNA, a sygnał fluorescencyjny jest stale wzmacniany.Metoda barwienia stosowana jest głównie w badaniach naukowych.
PS: Bądź ostrożny podczas przeprowadzania eksperymentu, barwnik musi być połączony z ludzkim DNA, uważaj, aby zmienić go w osobę fluorescencyjną.
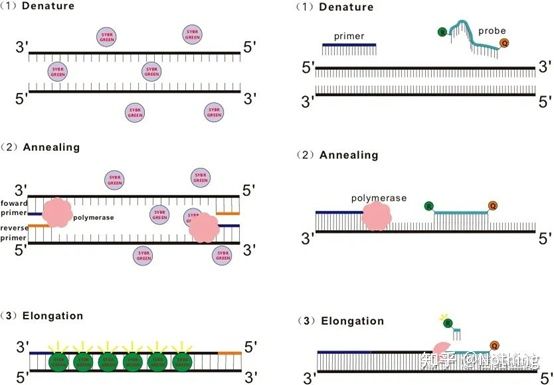
Metoda barwnika (po lewej) Metoda sondy (po prawej)
PS: Bądź ostrożny podczas przeprowadzania eksperymentu, barwnik musi być połączony z ludzkim DNA, uważaj, aby zmienić go w osobę fluorescencyjną.
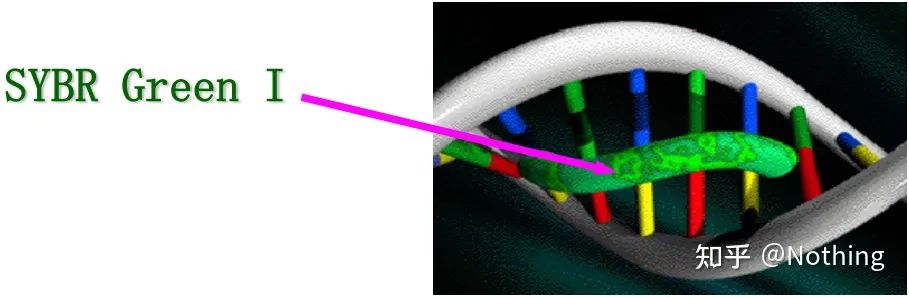
SYBR Green Ⅰ wiąże się z mniejszym rowkiem DNA
Metoda sondySonda Taqman jest najczęściej stosowaną sondą do hydrolizy.Na końcu 5' sondy znajduje się grupa fluorescencyjna, zwykle FAM, a sama sonda jest sekwencją komplementarną do docelowego genu.Na końcu 3' znajduje się grupa gasząca fluorescencję.Zgodnie z zasadą rezonansowego transferu energii fluorescencji (Förster rezonansowy transfer energii, FRET), kiedy reporterowa grupa fluorescencyjna (cząsteczka donorowa fluorescencyjna) i wygaszająca grupa fluorescencyjna (cząsteczka fluorescencyjna akceptorowa) są wzbudzone. Kiedy widma nakładają się, a odległość jest bardzo mała (7-10nm), wzbudzenie cząsteczki donorowej może indukować fluorescencję cząsteczki akceptorowej, podczas gdy autofluorescencja jest osłabiona.Dlatego na początku reakcji PCR, gdy sonda jest wolna i nienaruszona w układzie, reporterowa grupa fluorescencyjna nie będzie emitować fluorescencji.Podczas hybrydyzacji starter i sonda wiążą się z matrycą.Podczas fazy wydłużania polimeraza w sposób ciągły syntetyzuje nowe łańcuchy.Polimeraza DNA ma aktywność egzonukleazy 5′-3′.Po dotarciu do sondy polimeraza DNA zhydrolizuje sondę z matrycy, oddzieli reporterową grupę fluorescencyjną od wygaszającej grupy fluorescencyjnej i uwolni sygnał fluorescencyjny.Ponieważ istnieje relacja jeden do jednego między sondą a szablonem, metoda sondy przewyższa metodę barwnika pod względem dokładności i czułości testu.Metoda sondy stosowana jest głównie w diagnostyce.
P: Co to jest bezwzględna kwantyfikacja?Co to jest względna kwantyfikacja?
Odpowiedź: Bezwzględna ocena ilościowa odnosi się do obliczenia początkowej liczby kopii próbki do zbadania metodą qPCR, np. ile wirusów HBV znajduje się w 1 ml krwi.Wynikiem uzyskanym przez względną ocenę ilościową jest zmiana ilości docelowego genu w określonej próbce w stosunku do innej próbki referencyjnej, a ekspresja genu jest regulowana w górę lub w dół.
P: Czy ilość ekstrakcji RNA, wydajność odwrotnej transkrypcji i wydajność amplifikacji wpłyną na wyniki eksperymentu?
P: Czy przechowywanie próbek, odczynniki do ekstrakcji, odczynniki do odwrotnej transkrypcji i materiały eksploatacyjne przepuszczające światło wpłyną na wyniki eksperymentu?
P: Jaka metoda może poprawić dane eksperymentalne?
Jeśli chodzi o te kwestie, opiszemy je szczegółowo w sekcjach zaawansowanych i zaawansowanych poniżej.
2. Zaawansowana wiedza
Jeśli chodzi o fluorescencyjny ilościowy PCR w czasie rzeczywistym, musimy zdać sobie sprawę z rzeczywistości, że każdego roku publikowane są tysiące artykułów naukowych, wśród których technologia fluorescencyjnego ilościowego PCR nie jest małą liczbą.
Jeśli nie ma wspólnego standardu pomiaru fluorescencyjnego ilościowego eksperymentu PCR, wyniki mogą się znacznie różnić.W przypadku tego samego genu tego samego gatunku, z tą samą metodą przetwarzania, wyniki wykrywania również będą się znacznie różnić, a spóźnialskim trudno będzie powtórzyć te same wyniki.Ty Nikt nie wie, co jest dobre, a co złe.
Czy to oznacza, że fluorescencyjny ilościowy PCR jest technologią oszukańczą lub zawodną?Nie, to dlatego, że fluorescencyjny ilościowy PCR jest bardziej czuły i dokładniejszy, a niewielka niewłaściwa operacja da zupełnie odwrotne wyniki.Mała strata jest tysiące mil stąd.Autor artykułu może być wielokrotnie torturowany przez recenzentów.Jednocześnie recenzenci czasopisma są również trudni do wyboru spośród różnych wyników eksperymentów.
Podsumowując, wskazuje na brak konsensusu w eksperymentach PCR w czasie rzeczywistym.W tym celu starsi naukowcy z branży zaczęli formułować normy,wymaganie od autorów podania pewnych niezbędnych szczegółów dotyczących eksperymentów i przetwarzania danych (w tym niezbędnych danych) w artykule, aby spełnić te standardy.
Recenzenci mogą ocenić jakość eksperymentu, czytając te szczegóły;przyszli czytelnicy mogą również użyć tego do powtórzenia eksperymentu lub ulepszenia eksperymentu.Wtedy uzyskane w ten sposób wyniki eksperymentów są pełne informacji, wysokiej jakości i użyteczne.
MIBBI (minimalne informacje do badań biologicznych i biomedycznych -http://www.mibbi.org) powstała.MIBBI to projekt, który zapewnia standardy dla eksperymentów.Jest publikowany w naturze.Ten projekt jest ukierunkowany na różne eksperymenty biologiczne, w tym biologię komórki, mikromacierze, qPCR, o których teraz będziemy rozmawiać itp., i przewiduje każdy typ eksperymentu przy przesyłaniu manuskryptów.Informacje te powinny być podawane przez cały czas.
W projekcie MIBBI znajdują się dwa artykuły związane z fluorescencyjnym ilościowym PCR, a mianowicie:
·RDML (Real-Time PCR Data Markup Language) – ustrukturyzowany język i przewodnik raportowania ilościowych danych PCR w czasie rzeczywistym;
·MIQE (Minimum Information for Publication of Quantitative Real-Time PCR Experiments) – minimum informacji do publikowania artykułów na temat ilościowych eksperymentów PCR w czasie rzeczywistym.
Najpierw porozmawiajmy o RDML, specyfikacji terminologicznej.
Jeśli nie ma standardowej definicji wszystkiego, nie da się kontynuować dyskusji, dlatego wyjaśnienie terminów jest tak ważne na egzaminie.
Terminologia stosowana w eksperymencie fluorescencyjnej ilościowej reakcji PCR obejmuje następującą treść.Firma QIAGEN przygotowała dla nas najlepsze podsumowanie.Wszystkie poniższe są suchedobra .
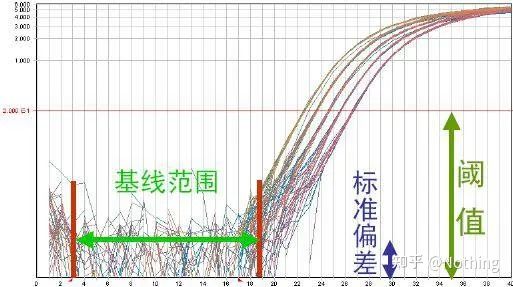
Krzywa amplifikacji
Krzywa amplifikacji odnosi się do krzywej wykonanej podczas procesu PCR, z numerem cyklu jako odciętą i intensywnością fluorescencji w czasie rzeczywistym podczas reakcji jako rzędną.
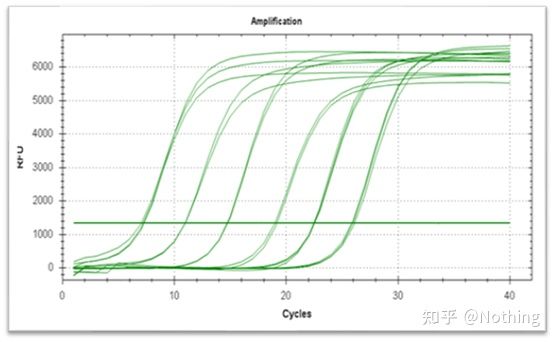
Doskonała krzywa amplifikacji powinna mieć następujące cechy: linia podstawowa jest płaska lub nieznacznie obniżona i nie ma wyraźnej tendencji wzrostowej;punkt przegięcia krzywej jest wyraźny, a nachylenie fazy wykładniczej jest proporcjonalne do wydajności amplifikacji.Im większe nachylenie, tym wyższa wydajność wzmocnienia;ogólna krzywa amplifikacji Równoległość jest dobra, co wskazuje, że wydajność amplifikacji każdej lampy jest podobna;wykładnicza faza krzywej amplifikacji próbek o niskim stężeniu jest oczywista.
Linia bazowa (Linia bazowa)
Linią bazową jest poziom hałasu wczesnego cyklu, zwykle mierzona między 3 a 15 cyklem, ponieważ wzrost wartości fluorescencji spowodowany przez produkt amplifikacji nie może być wykryty w tym okresie.Liczba cykli wykorzystanych do obliczenia linii podstawowej może być różna i może wymagać zmniejszenia, jeśli stosuje się duże ilości matrycy lub jeśli poziom ekspresji docelowego genu jest wysoki.
Ustawienie linii bazowej wymaga przejrzenia danych fluorescencji z krzywej amplifikacji liniowości.Linia podstawowa jest ustawiona tak, że wzrost krzywej amplifikacji zaczyna się od numeru cyklu większego niż górna liczba cyklu linii podstawowej.Linie bazowe należy ustalać indywidualnie dla każdej sekwencji docelowej.Średnie wartości fluorescencji wykryte we wczesnych cyklach należy odjąć od wartości fluorescencji uzyskanych w zamplifikowanych produktach.Najnowsze wersje różnych programów Real-Time PCR umożliwiają automatyczną optymalizację ustawień podstawowych dla poszczególnych próbek.
Podczas kilku pierwszych cykli reakcji amplifikacji PCR sygnał fluorescencji nie zmienia się zbytnio.Zbliżanie się do linii prostej nazywa się linią podstawową, ale jeśli przyjrzymy się bliżej kilku pierwszym cyklom, zobaczymy, że w obrębie linii podstawowej dzieje się to, co dzieje się na poniższym obrazku.
Tło Tło odnosi się do
niespecyficzna wartość fluorescencji w reakcji.Na przykład: niewydajne wygaszanie fluorescencji;lub duża liczba dwuniciowych matryc DNA dzięki zastosowaniu SYBR Green.Składowe tła sygnału są matematycznie usuwane przez algorytm oprogramowania Real-Time PCR.
Sygnał reporterski
Sygnał reporterowy odnosi się do sygnału fluorescencyjnego generowanego przez SYBR Green lub znakowane fluorescencyjnie sondy specyficzne dla sekwencji podczas reakcji PCR w czasie rzeczywistym.
Znormalizowany sygnał reportera (RN)
RN odnosi się do intensywności fluorescencji barwnika reporterowego podzielonej przez intensywność fluorescencji pasywnego barwnika odniesienia mierzoną w każdym cyklu.
Pasywny barwnik referencyjny
W niektórych reakcjach PCR w czasie rzeczywistym,barwnik fluorescencyjny ROX jest używany jako wewnętrzne odniesienie do normalizacji sygnału fluorescencyjnego.Koryguje odchylenia spowodowane niedokładnym pipetowaniem, pozycją dołków i fluktuacjami fluorescencji dla poszczególnych dołków.

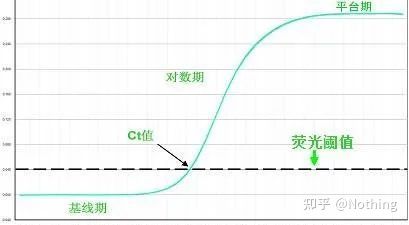
Próg fluorescencji (próg)
dostosowano powyżej wartości tła i znacznie poniżej wartości plateau krzywej amplifikacji.Musi leżeć w liniowym obszarze krzywej amplifikacji, reprezentującym logarytmiczno-liniowy zakres wykrywania PCR.Progi powinny być ustawione w widoku krzywej logarytmicznej amplifikacji, tak aby logarytmiczno-liniowa faza PCR była łatwo identyfikowalna.Jeśli w PCR w czasie rzeczywistym występuje wiele genów docelowych, dla każdego celu należy ustawić próg.Ogólnie, sygnał fluorescencji z pierwszych 15 cykli reakcji PCR jest używany jako sygnał tła fluorescencji, a próg fluorescencji jest 10-krotnością odchylenia standardowego sygnału fluorescencji z pierwszych 3 do 15 cykli PCR, a próg fluorescencji jest ustalany w wykładniczej fazie amplifikacji PCR.Ogólnie rzecz biorąc, każdy instrument ma ustawiony próg fluorescencji przed użyciem.
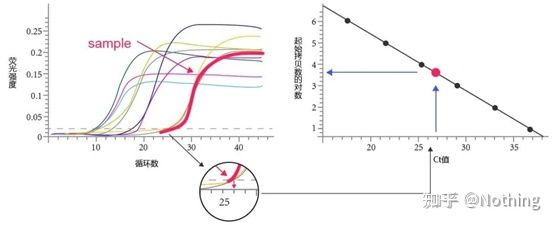
Próg cyklu (CT) lub punkt przecięcia (CP)
Cykl, w którym krzywa amplifikacji przekracza próg (tj. punkt, w którym wykrywanie fluorescencji znacznie wzrasta).CT może być ułamkiem i można obliczyć ilość szablonu wyjściowego.Wartość CT reprezentuje liczbę cykli występujących, gdy sygnał fluorescencyjny w każdej probówce do reakcji PCR osiąga ustawiony próg.Istnieje liniowa zależność między wartością CT każdego szablonu a logarytmem początkowej liczby kopii szablonu,im wyższa początkowa liczba kopii, tym mniejsza wartość CT i odwrotnie.Krzywą wzorcową można wykonać przy użyciu wzorca o znanej początkowej liczbie kopii, gdzie odcięta reprezentuje wartość CT, a rzędna reprezentuje logarytm początkowej liczby kopii.Dlatego dopóki uzyskana zostanie wartość CT nieznanej próbki, początkowa liczba kopii próbki może zostać obliczona z krzywej wzorcowej.
Wartość ΔCT
Wartość ΔCT opisujeróżnica między docelowym genem a odpowiednią wartością CT endogennego genu referencyjnego, taki jak gen porządkujący, i służy do normalizacji ilości użytego szablonu:
⇒ΔCT = CT (gen docelowy) – CT (endogenny gen referencyjny)
Wartość ΔΔCT
Wartość ΔΔCT opisuje różnicę między średnią wartością ΔΔCT badanej próbki (np. komórek stymulowanych) a średnią wartością ΔΔCT próbki referencyjnej (np. komórek niestymulowanych).Próbka referencyjna jest również nazywana próbką kalibracyjną, a wszystkie inne próbki są do niej normalizowane w celu względnej oceny ilościowej:
⇒ΔΔCT = średnia ΔCT (próbka będąca przedmiotem zainteresowania) – średnia ΔCT (próbka referencyjna)
Endogenne geny referencyjne (endogenne geny referencyjne)
Poziomy ekspresji endogennych genów referencyjnych, takich jak geny porządkowe (geny porządkowe), nie różnią się między próbkami.Porównanie wartości CT genu referencyjnego z genem docelowym umożliwia normalizację poziomu ekspresji genu docelowego do ilości wejściowego RNA lub cDNA (patrz sekcja dotycząca wartości ΔCT powyżej).
Wewnętrzne geny referencyjne poprawne dlamożliwą degradację RNA lub obecność inhibitorów enzymów w próbkach RNA, a także różnice w zawartości RNA, wydajność odwrotnej transkrypcji, odzyskiwanie kwasów nukleinowych i obchodzenie się z próbkami.Aby wybrać optymalny gen (geny) odniesienia, zmodyfikowaliśmy algorytm, aby umożliwić mu wybór optymalnego odniesienia w zależności od ustawień eksperymentalnych.
Kontrola wewnętrzna
Sekwencja kontrolna, która jest amplifikowana w tej samej reakcji co sekwencja docelowa i sondowana inną sondą (tj. przeprowadzając dupleksowy PCR).Kontrole wewnętrzne są często używane do wykluczenia nieudanych amplifikacji, na przykład gdy sekwencja docelowa nie została wykryta.
Próbka kalibracji
Próbka referencyjna (na przykład oczyszczony RNA z linii komórkowej lub tkanki) stosowana we względnej ocenie ilościowej w celu porównania wszystkich innych próbek w celu określenia względnego poziomu ekspresji genu.Próbką kalibracyjną może być dowolna próbka, ale zazwyczaj jest to próbka kontrolna (na przykład próbka nietraktowana lub próbka z czasu zerowego eksperymentu).
Kontrole pozytywne
użyj reakcji kontrolnych zznane ilości szablonu.Kontrole pozytywne są często używane do sprawdzania, czy zestaw starterów lub zestaw starter-sonda działa prawidłowo i czy reakcja jest prawidłowo ustawiona.
Brak kontroli szablonu (NTC)
Reakcja kontrolna zawierająca wszystkie niezbędne składniki reakcji amplifikacji z wyjątkiem matrycy, którą zwykle zastępuje się wodą.Zastosowanie NTC może wykryć zanieczyszczenie spowodowane zanieczyszczeniem odczynnikiem lub obcym DNA, zapewniając w ten sposób autentyczność i wiarygodność danych wykrywania.Amplifikacja kontroli NTC wskazuje na zanieczyszczenie.
Brak kontroli RT (NRT)
Proces ekstrakcji RNA może zawierać pozostałości genomowego DNA, które jest niezwykle szkodliwe i jest winowajcą wpływającym na jakość danych oraz naturalnym wrogiem qPCR, dlatego podczas projektowania eksperymentów należy go zaprojektować tak, aby tylko wzmacniał wykrywanie RNA.Istnieją dwa sposoby, jeden to zaprojektowanie starterów w poprzek intronów, drugi to całkowite usunięcie DNA, który jest lepszy, co zostanie omówione później.Kontrola NTR to magiczne lustro do wykrywania zanieczyszczenia DNA.Jeśli występuje wzmocnienie, oznacza to zanieczyszczenie.
Normy
Standardy to próbki o znanym stężeniu lub liczbie kopii, które są używane do konstruowania krzywej wzorcowej.W celu zapewnienia stabilności standardu fragment genu jest zwykle klonowany do plazmidu i używany jako wzorzec.
Krzywa standardowa
jest zwykle rozcieńczany do co najmniej 5 gradientów stężeń z produktem wzorcowym zgodnie ze współczynnikiem podwojenia, a 5 punktów jest rysowanych we współrzędnych wartości CT i liczby kopii, a punkty są łączone w celu utworzenia linii w celu wygenerowania krzywej wzorcowej.Dla każdej krzywej wzorcowej należy sprawdzić jej ważność.Wartość nachylenia mieści się w zakresie od –3,3 do –3,8, a każde stężenie wykonuje się trzykrotnie.Punkty, które znacznie różnią się od innych punktów, należy odrzucić.Wartość CT badanej próbki umieszcza się na krzywej wzorcowej i można obliczyć poziom ekspresji badanej próbki.
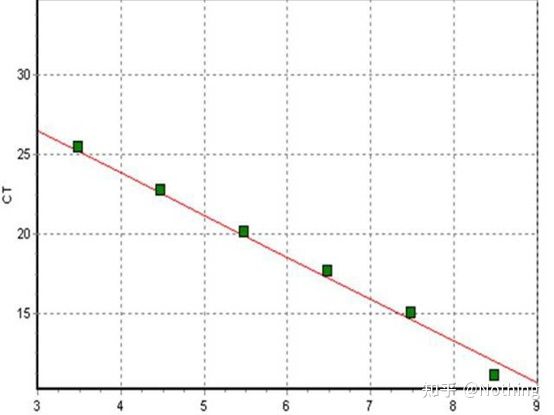
Wartość CT badanej próbki umieszcza się na krzywej wzorcowej i można obliczyć początkową liczbę kopii badanej próbki.
Wydajność i nachylenie
Nachylenie krzywej wzorcowej przedstawia wydajność PCR w czasie rzeczywistym.
·Nachylenie -3,322 wskazuje, że wydajność amplifikacji PCR wynosi 1 lub 100%, a ilość produktu PCR podwaja się w każdym cyklu.
·Nachylenie mniejsze niż –3,322 (np. –3,8) wskazuje na wydajność PCR
·Nachylenie większe niż –3,322 (np. –3,0) wskazuje, że wydajność PCR wydaje się być większa niż 100%, co jest ciekawe, w jaki sposób jeden cykl PCR może generować więcej niż dwukrotnie zamplifikowany produkt?Taka sytuacja występuje w nieliniowej fazie reakcji PCR, to znaczy występuje duża ilość amplifikacji niespecyficznej.
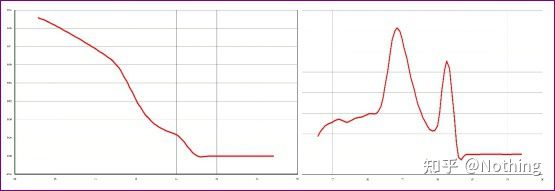
krzywa topnienia
Po zakończeniu amplifikacji qPCR produkt PCR jest podgrzewany.Wraz ze wzrostem temperatury dwuniciowy produkt amplifikacji stopniowo topi się, co powoduje spadek intensywności fluorescencji.Po osiągnięciu określonej temperatury (Tm) duża liczba produktów ulegnie stopieniu.Fluorescencja gwałtownie spada.Różne produkty PCR mają różne wartości Tm i różne temperatury topnienia, dzięki czemu można określić specyficzność PCR.
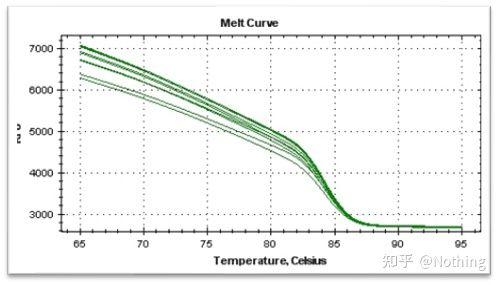
Krzywa topnienia (krzywa pochodna)
Krzywa topnienia jest wyznaczana w celu utworzenia mapy pików, która może bardziej intuicyjnie przedstawiać sytuację fragmentów produktu PCR.Ponieważ temperatura topnienia jest wartością Tm fragmentu DNA, można ocenić niektóre parametry wpływające na wartość Tm fragmentu DNA, takie jak rozmiar fragmentu, zawartość GC itp. Ogólnie rzecz biorąc, zgodnie z naszymi zasadami projektowania starterów,długość amplifikowanego produktu mieści się w zakresie 80-300 pz, więc temperatura topnienia powinna wynosić od 80°C do 90°C.
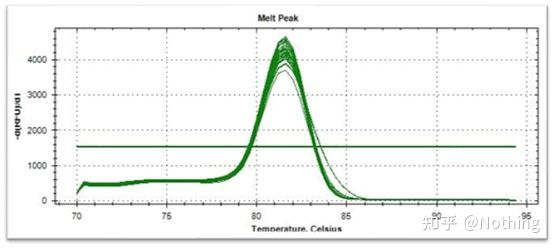
Interpretacja krzywej topnienia: Jeśli jedyny główny pik pojawia się między 80°C-90°C, oznacza to, że fluorescencyjny ilościowy PCR jest doskonały;jeśli główny pik pojawia się między 80°C-90°C, a różne piki pojawiają się poniżej 80°C, zasadniczo bierze się pod uwagę dimer startera.Możesz spróbować zwiększyć temperaturę wyżarzania, aby go rozwiązać;jeśli główny pik pojawia się między 80°C-90°C, a inny pik pojawia się ponownie, gdy temperatura wzrasta, zasadniczo uważa się, że doszło do zanieczyszczenia DNA i DNA należy usunąć na początkowym etapie eksperymentu.
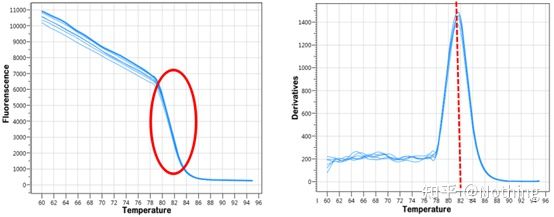
Oczywiście wciąż zdarzają się sytuacje odbiegające od normy, które zostaną omówione po kolei poniżej.
3. Zaawansowana wiedza
Aby wykonać qPCR, muszę powiedzieć MIQE,Minimalne informacjeza publikacjęIlościowyPCR w czasie rzeczywistymEksperymenty — minimum informacji do publikowania artykułów na temat ilościowego PCR w czasie rzeczywistymeksperymenty.Aby ułatwić wszystkim zrozumienie, uprościmy kluczową treść.
Możesz wyszukać oryginalny tekst MIQE w Internecie, a najważniejsze jest to, że określachecklista danych, które należy podać przy publikacji artykułu .
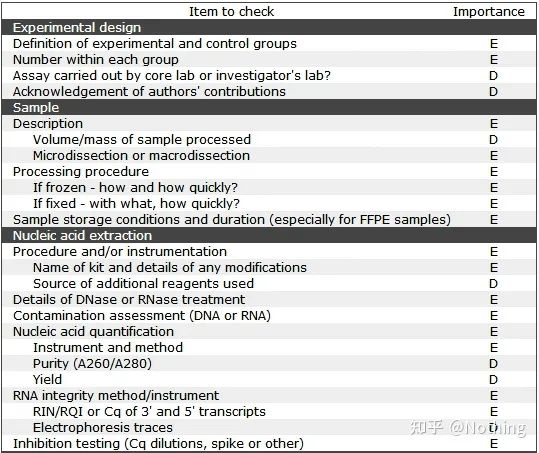
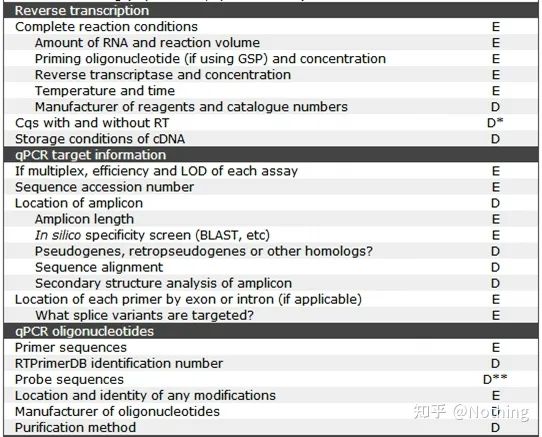
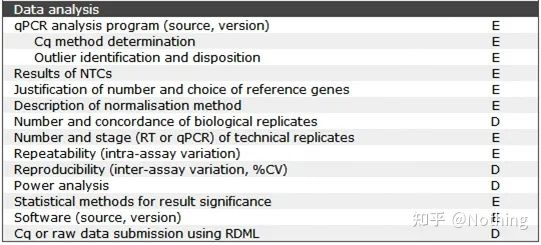
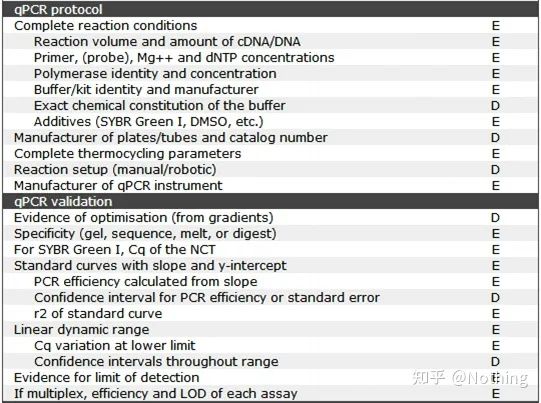
Recenzenci mogą ocenić jakość eksperymentu, czytając te szczegóły;przyszli czytelnicy mogą również użyć tego do powtórzenia lub ulepszenia eksperymentu.
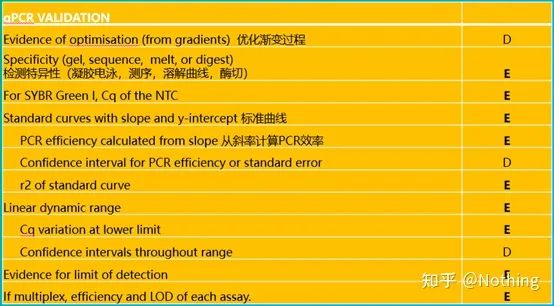
Warto zauważyć, że na tej liście ważność każdej listy jest oznaczona odpowiednio literą E lub D.Co to znaczy?E: podstawowe informacje (należy podać);D: pożądane informacje (podaj jak najwięcej).
MIQE (1) — projekt eksperymentalny
Wielu szumowin, którzy zakończyli obronę po ukończeniu studiów podyplomowych, nie będzie wiedziało, jak samodzielnie zaprojektować eksperyment, otworzyć zeszyty i zrobić to, co każe im nauczyciel.W rezultacie projekt eksperymentalny nie był rygorystyczny, a redakcja magazynu powiedziała, że chce wymyślić to i tamto zdjęcie, więc zrobili to w oszołomieniu.Tak się robi skurwysyny!

Bliżej domu, pierwszą zasadą eksperymentu jest ustalenierygor logiki eksperymentalnej.Najbardziej fundamentalną rzeczą jest projekt eksperymentu, a najważniejszą rzeczą w projekcie eksperymentu jest to, jak ustawić próbkę docelową, próbkę referencyjną (kontrolną) i liczbę powtórzeń, tak aby dane eksperymentalne można było odwoływać, porównywać i przekonywać.
Próbka docelowaodnosi się do próbki, która wymaga od nas wykrycia docelowego genu po pewnym leczeniu.Próbka referencyjnato próbka bez żadnej obróbki, która w biologii jest często określana jako typ dziki.
Eksperymentalne powtórzeniasą bardzo ważne.Ogólnie rzecz biorąc, liczba perswazyjnych powtórzeń musi być większa niż trzy.Konieczne jest rozróżnienie, czym jest replikacja biologiczna, a czym jest replikacja techniczna.
Repliki biologiczne: Ten sam eksperyment weryfikacyjny przeprowadzony z różnymi materiałami (czas, rośliny, partie, płytki reakcyjne).

Duplikacja biologiczna
Weźmy jako przykład traktowanie pieprzu pestycydami.Chcemy spryskać pestycydami trzy rośliny ABC, a następnie trzy rośliny ABC są trzema powtórzeniami biologicznymi i są tym samym eksperymentem weryfikacyjnym przeprowadzonym z różnymi materiałami.Ale jako eksperyment zdecydowanie potrzebna jest kontrola, więc możemy opryskać jedną z gałęzi rośliny A, aby utworzyć eksperymentalną grupę rośliny A, i nie opryskać innych gałęzi rośliny A, aby utworzyć grupę kontrolną.Zrób to samo dla B i C.
Repliki techniczne (repliki techniczne): Jest to powtarzany eksperyment mający na celu uniknięcie błędów spowodowanych działaniem, które w rzeczywistości jest zduplikowanym otworem zawartym w tym samym materiale.Zarówno zabiegi, jak i kontrole muszą mieć powtórzone ustawienia (minimum trzy) genu docelowego i wewnętrznego genu referencyjnego.
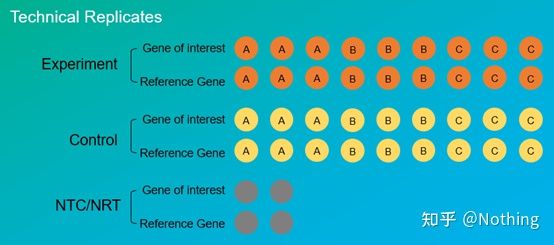
Powtórzenie techniczne
Weźmy ponownie za przykład paprykę potraktowaną pestycydami.Dla grupy eksperymentalnej rośliny A wykonaliśmy trzy otwory PCR o wartości 1, 2 i 3 odpowiednio dla jej genu docelowego i wewnętrznego genu odniesienia, aby wziąć średnią po wykryciu.W celu zwalczania roślin Grupy A są również traktowane w ten sam sposób.Podobnie wykonaj ten sam zabieg dla roślin B i C.To jest techniczna powtórka.
Warto to zauważyćto, co wchodzi do statystyki, to powtórzenie biologiczne, a powtórzenie techniczne ma na celu sprawdzenie, czy w procesie eksperymentalnym występują jakiekolwiek zjawiska losowe, aby wyniki eksperymentalne były wiarygodne, to znaczy, aby uniknąć błędów, biorąc ich średnią, jak często mówimy.
Kontrole negatywne — NTC i NRT
NTC (kontrola bez szablonu), kontrola bez szablonu, służy do sprawdzenia, czy materiał doświadczalny jest zanieczyszczony.Ogólnie woda jest używana jako szablon.Jeśli występuje reakcja fluorescencyjna, oznacza to, że w laboratorium doszło do zanieczyszczenia kwasem nukleinowym.
Zanieczyszczenia te pochodzą z: zanieczyszczonej wody, niewykwalifikowanych odczynników zawierających endogenne DNA, zanieczyszczenia starterów, zanieczyszczenia sprzętu laboratoryjnego, zanieczyszczenia aerozolem itp., konieczność stosowania zmiataczy RNaz i inhibitorów RNaz.Zanieczyszczenia aerozolowe są najtrudniejsze do wykrycia.Wyobraź sobie, że twoje laboratorium jest jak smog, z różnymi kwasami nukleinowymi zawieszonymi w powietrzu.

NRT (bez odwrotnej transkryptazy), kontrola bez odwrotnej transkrypcji, jest RNA nie podlegającym odwrotnej transkrypcji jako kontrola negatywna, która jest kontrolą reszty gDNA.
Podczas ekspresji genów ilość RNA jest wykrywana przez wykrywanie ilości cDNA po odwrotnej transkrypcji.Jeśli podczas oczyszczania RNA będzie obecna pozostałość gDNA, spowoduje to błędy w wynikach eksperymentalnych, ponieważ faktycznie otrzymane wyniki to gDNA i cDNA.Na poziomie agregatów, nie tylko cDNA, gDNA musi zostać całkowicie usunięty podczas ekstrakcji RNA.
MIQE (2) — przykładowe informacje
Tak zwane informacje o próbce oznaczają, że publikując artykuł o qPCR, musimy jasno wyjaśnić informacje o próbce, co jest nieodzowną częścią artykułu.Podobnie, gdy przetwarzamy próbki, musimy również regulować nasze własne działania, aby zapewnić ważność próbek.

Opis próbki jest tylko wynikiem i powinniśmy zwrócić większą uwagę na materiały pobrane podczas całego eksperymentu.
Dobór materiałów doświadczalnych
Próbki krwi – wybierz świeżą krew, nie więcej niż 4 godziny.Próbki komórek – wybierz pobieranie świeżych komórek w okresie intensywnego wzrostu.Tkanka zwierzęca — wybierz świeżą, silnie rosnącą tkankę.Tkanka roślinna – Wybierz świeżą, młodą tkankę.
Na pewno zauważyliście, że w tych kilku zdaniach jest słowo klucz: świeży .
W przypadku powyższych próbek najlepszym, ekonomicznym i stabilnym zestawem na rynku jest zestaw Foregene, który może szybko i łatwo wyodrębnić ich DNA i RNA.
Zestaw do izolacji całkowitego RNA komórek
Zestaw do izolacji całkowitego RNA zwierząt
Zestaw do izolacji całkowitego RNA roślin
Zestaw do izolacji całkowitego RNA roślin Plus
Przechowywanie materiałów doświadczalnych
Ogólnie rzecz biorąc, nie zalecamy przechowywania próbek, jeśli pozwalają na to warunki.Jednak jest wielu przyjaciół, którzy nie mogą przeprowadzać eksperymentów natychmiast po pobraniu próbek, a niektórzy nawet muszą nosić zbiorniki z ciekłym azotem na pole w celu pobrania próbek.
Dla tego rodzaju ciężko pracującego przyjaciela mogę tylko powiedzieć, że nie rozumiesz materiałów eksploatacyjnych odczynników.Obecnie wiele firm produkujących materiały eksploatacyjne do odczynników produkuje odczynniki, które mogą przechowywać próbki RNA w temperaturze pokojowej i można z nich korzystać.Konwencjonalną metodą przechowywania jest przechowywanie ciekłego azotu przy użyciu małego, łatwego do przenoszenia zbiornika z ciekłym azotem.Po przywiezieniu próbki do laboratorium przechowuj ją w lodówce -80°C.

W przypadku eksperymentów z udziałem RNA należy przestrzegać zasady sześciu słów:niska temperatura , brak enzymów ,Iszybko .
Pojęcie niskiej temperatury jest łatwe do zrozumienia;bez enzymów RNaza jest wszędzie na świecie, w którym żyjemy (inaczej zostałbyś zabity przez HIV), więc jak unikać RNazy podczas przeprowadzania eksperymentów, to bardzo ważna koncepcja;szybko,Na świecie nie ma Kung Fu, którego nie można złamać, nie można złamać tylko szybkości.
Dlatego w pewnym sensie im krótszy czas ekstrakcji, tym lepszy zestaw.DlaczegoForegeneKit kładą nacisk na szybkość, bo dobrze ją znają.
PS: Niektóre dziewczyny wykonują eksperymenty bardzo ostrożnie, ale po kilku latach pracy nie są tak dobre jak wsad.Czują, że Bóg jest niesprawiedliwy, narzeka na innych i szuka życia.Prawdę mówiąc, nie rozumiała tego.Nie chronił dobrze RNA, a gracz wykonujący wsad był zwinny.Kiedy przeprowadzał eksperyment, myślał, że zakończy wsad trzykrotnie, pięciokrotnie i dwoma podziałami, ale eksperyment wykonał dobrze.
Notatka: Wolniej, większe szanse na inwazję RNazy.Jak trenować, aby być szybkim?Nie ma sposobu, po prostu ćwicz więcej.
W przypadku różnych eksperymentów i różnych próbek nadal konieczne jest przeczytanie większej ilości literatury i wybranie odpowiedniej metody przetwarzania.W przypadku procesu pobierania i przechowywania próbek MIQE wymaga, aby było to wyraźnie napisane w artykule, tak aby recenzenci mogli sprawdzić wiarygodność artykułu, a także dla oszołomionych młodych ludzi wygodnie jest powtórzyć eksperyment.
Chociaż eksperymenty biologiczne są trudne, są wysokiej klasy.Jeśli nie będziesz ostrożny, możesz obalić świat.Na przykład przekształcenie SARS w kryzys biochemiczny lub zrobienie hybrydowego ryżu, aby uratować 1,3 miliarda ludzi.Poniższe zdjęcie przedstawia eksperyment chemiczny, powinieneś zrozumieć, jak bardzo jesteś dumny ze swoich badań, patrząc tylko na jego wygląd przypominający penisa.Zapomnij o tym, nie oczerniaj go.

MIQE (3) – ekstrakcja kwasu nukleinowego.
Ekstrakcja kwasu nukleinowego to wielkie wydarzenie, a wszystkie eksperymenty biologii molekularnej rozpoczynają się od ekstrakcji kwasu nukleinowego.Przede wszystkim skopiujmy zawartość MIQE na temat ekstrakcji kwasu nukleinowego.
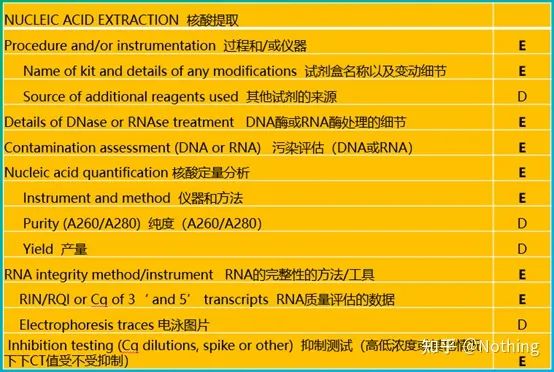
Patrząc na tę formę, nie możesz pozostać na powierzchni.Forma jest dogmatem.Aby być prymusem, musisz zapytać dlaczego.Zasadnicza zawartość tej tabeli to: Pościgczystość, integralność, spójność i ilość ekstrakcji RNA .
Pierwsza część wwprocesem lub instrumentem jest etap ekstrakcji kwasu nukleinowego.Jeśli używasz automatycznego ekstraktora kwasów nukleinowych do ekstrakcji (zaawansowany, skontaktuj się ze mną w celu zakupu), musisz podać nazwę modelu instrumentu.

Nazwa zestawu i
jaki zestaw został użyty do zmiany szczegółów, jakie specjalne odczynniki zostały dodane lub jakie specjalne operacje zostały wykonane, należy jasno wyjaśnić, aby inni mogli łatwo powtórzyć Twój eksperyment.
Niektórzy ludzie dodają specjalne odczynniki podczas ekstrakcji specjalnych próbek, myśląc, że to ich tajna broń i nie mówcie o tym innym.Utrzymując to w tajemnicy, tracą również możliwość zabłyśnięcia Twojego artykułu.Nie bądź sprytny, musisz być bardziej uczciwy niż stary wiejski Zhang w badaniach naukowych, jeśli chcesz być sprytny, artykuł zrobi z ciebie głupka.
należy zapamiętać numer produktu zestawukiedy zamawiasz zestaw i piszesz artykuł.Na zestawie zazwyczaj znajdują się dwa numery: Cat – numer katalogowy (numer produktu, numer artykułu), Lot – numer partii produktu ( Służy do wskazania partii, z której pochodzi produkt).
Ponadto numer CAS jest często używany przy zamawianiu odczynników biochemicznych i będę go wspólnie popularyzował.Numer CAS to numer nadawany przez American Chemical Society każdemu nowemu lekowi chemicznemu.Ogólnie rzecz biorąc, trzy liczby są połączone myślnikiem.Numer CAS Rushui: 7732-18-5.Chemikalia często mają wiele aliasów, ale numer CAS jest unikalny.Zamawiając lek, możesz najpierw sprawdzić jego numer CAS.
Bliżej domu, dlaczego musimy jasno opisywać te rzeczy?W rzeczywistości ma to również na celu sprawdzenie jakości ekstrakcji RNA.Zastosowanie narzędzi i zestawów sprawi, że ekstrakcja RNA będzie bardziej spójna.Skala ekstrakcji zwykłych laboratoriów nie jest duża i można ją uzyskać za pomocą zestawów.
Szczegóły leczenia DNazą lub RNazą
Ważną kwestią fluorescencyjnej ilościowej PCR jest zapobieganie zanieczyszczeniu DNA i nie eksperymentowanie, jeśli występuje zanieczyszczenie.Dlatego konieczne jest podanie procesu użytego do przetworzenia DNA, aby wykazać, że DNA w procesie eksperymentalnym zostało całkowicie i całkowicie usunięte.reprezentowane przez schematyczny diagram.
Schematyczny diagram RNA i DNA
Ogólnie metoda usuwania DNA polega na potraktowaniu RNA DNazą po ekstrakcji.Są to jednak stosunkowo stare metody.Komercyjne zestawy do ekstrakcji RNA były w stanie usunąć DNA podczas procesu ekstrakcji bez dodawania DNazy.Na przykład seria zestawów od Foregene.
Notatka: Usuwanie DNA podczas ekstrakcji RNA to bardzo niebezpieczny miecz obosieczny, który wydłuży czas operacji ekstrakcji RNA i zwiększy ryzyko degradacji RNA.Zasadniczo jest to kompromis między wydajnością RNA a czystością.
Ponadto ilość DNazy dodanej do kolumny adsorpcyjnej na bazie krzemionki jest bardzo mała i aby osiągnąć efekt, należy użyć wysokiej jakości DNazy.Niezoptymalizowana DNaza nie może być trawiona szybko i całkowicie.Jest to test poziomu technicznego sprzedawcy.Oczywiście jest jeszcze więcej dziwnych handlarzy, którzy chwalą się, że DNA można usunąć bez DNazy.Można powiedzieć, że każdy, kto chwali się, że DNA można całkowicie usunąć bez DNazy, jest chuliganem.DNA jest stosunkowo stabilną dwuniciową strukturą i nie można go wymazać samą rozmową i śmiechem.
Ocena zanieczyszczenia
metoda oceny: detekcja elektroforetyczna, 1% agaroza, 6V/cm, 15min, ładowanie 1-3 ul
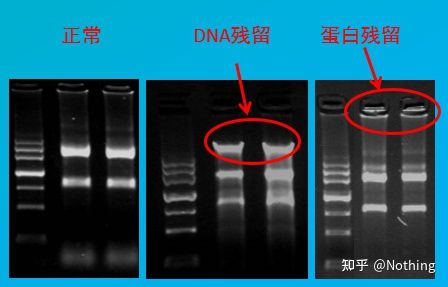
Analiza ilościowa kwasów nukleinowych
jest zwykle mierzona za pomocą spektrofotometru UV.Pozwólcie, że najpierw spopularyzuję znaczenie trzech wartości OD260, OD280 i OD230.
·OD260nm: Jest to długość fali absorpcji najwyższego piku absorpcji kwasu nukleinowego, a najlepiej zmierzona wartość mieści się w zakresie od 0,1 do 1,0.Jeśli nie, rozcieńczyć lub zatężyć próbkę tak, aby mieściła się w zakresie.
·OD280nm: Jest to długość fali absorpcji najwyższego piku absorpcji substancji białkowych i fenolowych.
·OD230nm: Jest to długość fali absorpcji najwyższego piku absorpcji węglowodanów.
Następnie porozmawiajmy o roli każdego wskaźnika.W przypadku A260 można go stosować do pomiaru wydajności kwasu nukleinowego.Gdy OD260=1, dsDNA=50μg/ml, ssDNA=37μg/ml, RNA=40μg/ml.
Aby uzyskać czystość, musimy spojrzeć na proporcje, które powszechnie widzimy: OD260/280 i OD260/230.
·Czyste DNA: OD260/280 jest w przybliżeniu równe 1,8.Gdy jest większy niż 1,9, wskazuje to na zanieczyszczenie RNA, a gdy jest mniejszy niż 1,6, wskazuje na zanieczyszczenie białkiem i fenolem.
·Czysty RNA: 1,7
·OD260/230: Niezależnie od tego, czy chodzi o DNA, czy RNA, wartość referencyjna wynosi 2,5.Gdy jest mniejszy niż 2,0, oznacza to zanieczyszczenie cukrem, solą i materią organiczną.
Integralność RNA
Bardzo ważne jest zmierzenie integralności RNA.Ogólnie rzecz biorąc, konieczne jest wykonanie eksperymentu w żelu do denaturacji RNA, aby sprawdzić, czy jasność między 28S a 18S RNA jest dwukrotna.Pojawienie się trzeciego prążka 5S oznacza, że RNA zaczęło się rozkładać, z wyjątkiem bezkręgowców.

Dane do oceny jakości RNA: Oprócz powyższych testów istnieją również bardziej zaawansowane testy przyrządowe pod względem integralności RNA, takie jak test integralności RQI systemu automatycznej elektroforezy Experion, który może wykryć, czy RNA ulega niewidocznej degradacji.
W badaniach naukowych fluorescencyjny ilościowy PCR polega na porównaniu genu docelowego z wewnętrznym genem referencyjnym.Dlatego w procesie konserwacji próbek RNA, ekstrakcji RNA itp. nadrzędnym celem jest zapewnienie integralności RNA.
Jak integralność RNA wpływa na równowagę między genem docelowym a wewnętrznym genem referencyjnym, można łatwo zrozumieć na podstawie poniższego rysunku.Degradacja doprowadzi do niekompletności genu, niezależnie od tego, czy jest to niekompletność wewnętrznego genu referencyjnego, czy niekompletność genu docelowego, będzie to miało ogromny wpływ na dane.

Schematyczny diagram genu docelowego i genu referencyjnego nie może być prawdziwy
Test hamowania (czy wartość CT jest tłumiona przy wysokim lub niskim stężeniu lub w innych warunkach)
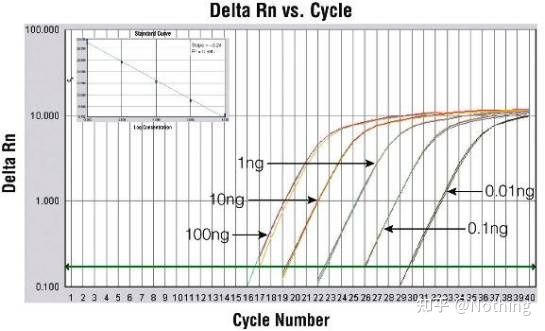
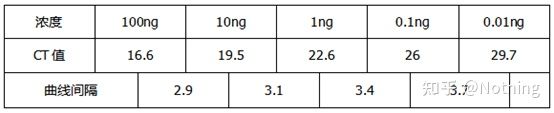
Biorąc ten rysunek jako przykład, wartości Ct pięciu krzywych są następujące.Rozkład wartości CT między krzywymi jest nierównomierny, a wartości Ct są opóźnione w przypadku wysokich i niskich stężeń, co ma miejsce w przypadku hamowania PCR.
Kluczowy punkt: W procesie ekstrakcji RNA musimy porzucić błędne przekonania i ustalić prawidłowe.
Błędny pomysł jest taki, że ekstrakcja RNA ma na celu tylko wydajność, myśląc, że im większa ilość uzyskanego RNA, tym lepiej.W rzeczywistości, kiedy dokonujemy oceny ilościowej, jeśli liczba genów nie jest bardzo duża, nie potrzebujemy dużo RNA.Ilość ekstrahowanego RNA jest więcej niż wystarczająca.
Prawidłowa koncepcja to:Ekstrakcja RNA powinna dążyć do czystości, integralności i spójności.Czystość może zapewnić, że późniejsza odwrotna transkrypcja nie zostanie zahamowana, a DNA nie wpłynie na dane.Integralność zapewnia równowagę sekwencji docelowych i wewnętrznych odniesień.Spójność zapewnia stabilne ładowanie próbki.
MIQE (4) – odwrotna transkrypcja
Nieporozumienie: dążenie do większej objętości próbki.
Prawidłowa koncepcja: Dążenie do spójności (stabilności), niezależnie od ilości załadowanego RNA, wydajność odwrotnej transkrypcji pozostaje stała, zapewniając, że różnice w cDNA mogą naprawdę odzwierciedlać różnice w mRNA.
Wyjaśniamy ten proces za pomocą schematu:
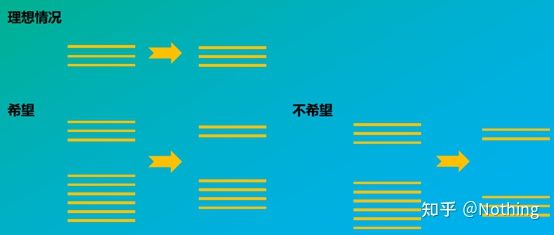
Schematyczny diagram wydajności odwrotnej transkrypcji nie może być prawdziwy
Przede wszystkim musimy zrozumieć różnicę między procesem odwrotnej transkrypcji a procesem PCR.PCR przechodzi wiele procesów ogrzewania i hybrydyzacji, a docelowy fragment rośnie wykładniczo;podczas gdy odwrotna transkrypcja nie ma tego procesu, możemy sobie wyobrazić, że odwrotna transkrypcja jest w rzeczywistości jeden do jednego Podczas procesu replikacji tyle fragmentów RNA
Ponieważ można uzyskać tyle kawałków informacji cDNA, należy to już zrozumieć, ponieważ duże i małe fragmenty zostały poddane odwrotnej transkrypcji i nie można skupić się na jednym fragmencie.A ponieważ ilość RNA jest stosunkowo niewielka, ilość otrzymanego cDNA jest również stosunkowo niewielka, w przeciwieństwie do PCR, który ma efekt amplifikacji, więc jest w zasadzie niemożliwy do wykrycia.
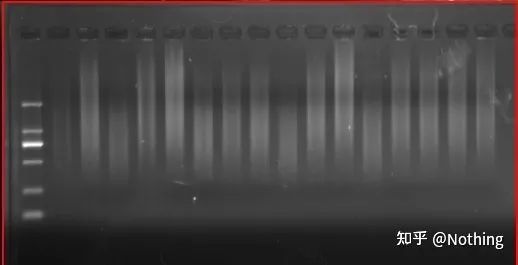
Wyniki elektroforezy cDNA
Po drugie, w idealnym przypadku odwrotna transkrypcja jest wykonywana jeden do jednego, ale żadna odwrotna transkryptaza z żadnej firmy nie może osiągnąć takiego efektu.Zasadniczo wydajność większości odwrotnych transkryptaz waha się między 30-50%.W takim przypadku wolelibyśmy raczej mieć stosunkowo stabilną wydajność odwrotnej transkrypcji, co chcemy zobaczyć na rysunku: 3 RNA otrzymują 2 cDNA, 6 RNA otrzymuje 4 cDNA, więc bez względu na to, ile próbki jest załadowane, wydajność odwrotnej transkrypcji jest względnie stabilna.Nie chcemy sytuacji, w której wydajność odwrotnej transkrypcji jest niestabilna, a wysoka koncentracja jest hamowana.
Jak więc sprawdzić, czy wydajność odwrotnej transkrypcji jest stabilna?Metoda jest bardzo prosta, wystarczy wykonać test porównawczy: jeden polega na odwrotnej transkrypcji do cDNA po dwukrotnym rozcieńczeniu RNA, a drugi to wykonanie dwukrotnego rozcieńczenia po odwrotnej transkrypcji na cDNA, a następnie wykonanie qPCR, aby zobaczyć otrzymane nachylenie Czy jest spójne.Jako prymus powinieneś zrozumieć to w kilka sekund.Jak pokazano niżej:
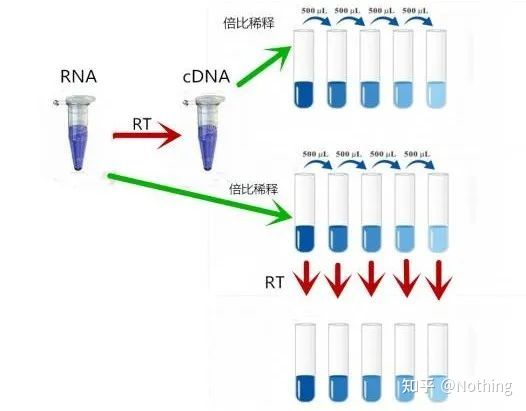
Rozcieńczenie RNA i cDNA w celu sprawdzenia, czy wydajność odwrotnej transkrypcji jest stabilna
Odwrotna transkryptaza i zestaw
W jaki sposób doskonały fluorescencyjny ilościowy PCR może mieć doskonałą odwrotną transkryptazę i zestaw.Odwrotna transkryptaza jest z grubsza podzielona na dwa typy w zależności od źródła, AMV lubM-MLV, a ich wydajność jest taka sama, jak pokazano w tabeli.
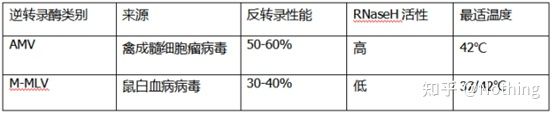
Aktywność RNazy H
RNaza H to rybonukleaza H, chińska nazwa to rybonukleaza H, która jest endorybonukleazą, która może specyficznie hydrolizować RNA w hybrydowym łańcuchu DNA-RNA.RNaza H nie może hydrolizować wiązań fosfodiestrowych w jednoniciowym lub dwuniciowym DNA lub RNA, to znaczy nie może trawić jednoniciowego lub dwuniciowego DNA lub RNA.Powszechnie stosowany w syntezie drugiej nici cDNA.
To dziwna rzecz.Mówimy, że odwrotna transkryptaza ma aktywność RNazy H, a nie, że odwrotna transkryptaza zawiera RNazę H, i może nie być możliwe oddzielenie RNazy H od odwrotnej transkryptazy, być może z powodu konformacji pewnych grup w odwrotnej transkryptazie. Ta aktywność jest spowodowana przez odwrotną transkryptazę.
Dlatego niezależnie od wyższej wydajności odwrotnej transkrypcji AMV, jego aktywność RNazy H zmniejsza wydajność cDNA.Oczywiście producenci odczynników stale optymalizują swoje produkty, aby w jak największym stopniu wyeliminować aktywność RNazy H w odwrotnej transkryptazie, aby zwiększyć wydajność cDNA.
Temperatura wyżarzania
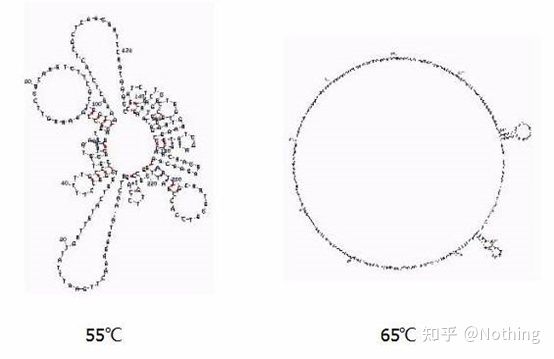
Struktura drugorzędowa RNA w różnych temperaturach
Zobacz rysunek powyżej, aby zobaczyć drugorzędową strukturę RNA w różnych temperaturach i użyj internetowego narzędzia mFold, aby określić drugorzędową strukturę docelowego fragmentu w określonych warunkach temperatury i stężenia soli.W temperaturze 55°C struktura drugorzędowa RNA jest nadal bardzo złożona, odwrotna transkryptaza nie może działać, a struktury drugorzędowej nie można całkowicie rozdzielić do temperatury 65°C, podczas gdy optymalna temperatura AMV i M-MLV jest znacznie niższa od tej temperatury.
co robić?Struktura drugorzędowa to komplementarne parowanie samej matrycy, co prowadzi do silnej konkurencji między starterem i odwrotną transkryptazą a matrycą, co skutkuje szeregiem problemów, takich jak niska E i słaba powtarzalność.
co robić?Temperaturę wyżarzania należy zwiększać tylko tak bardzo, jak to możliwe.
Wielu producentów odczynników ulepsza swoją odwrotną transkryptazę poprzez inżynierię genetyczną.Niektóre zwiększają temperaturę reakcji, na przykład Jifan i Aidelai, a niektóre usuwają grupę aktywną enzymu RNazy H, aby poprawić powinowactwo między enzymem a matrycą RNA.Wysokie powinowactwo może konkurencyjnie wycisnąć strukturę drugorzędową i płynnie czytać, a także znacznie poprawić wydajność odwrotnej transkrypcji.
Kluczowy punkt: Odwrotna transkrypcja jest ważniejsza niż dążenie do spójności wydajności odwrotnej transkrypcji (enzymy muszą być nie tylko wydajne, ale także stabilne), niż ilość załadowanej próbki, jeśli nie jest to ilościowa PCR fluorescencyjna na szczególnie dużą skalę, nie będzie to w ogóle możliwe.Wiele cDNA.
Różni producenci również poczynili pewne wysiłki w dążeniu do spójności.Na przykład większość firm oferuje teraz odwrotną transkrypcję jako standardowy zestaw do sprzedaży, co jest dobrym wyborem.
Na przykład zestawy RT Easy Series firmy Foregene:
RT Easy I (premiks główny do syntezy pierwszej nici cDNA)
MIQE (5) – informacja o genie docelowym

Powyższy rysunek wyjaśnia
1. To, czy ten gen jest skuteczny w powtarzanych eksperymentach, można ogólnie zweryfikować za pomocą powtarzanych eksperymentów.
2. Identyfikator genu, wiesz.
3. Długość genu, całkowita długość docelowego genu zdecydowanie nie stanowi problemu.Projektując startery, upewnij się, że długość amplikonu mieści się w przedziale 80-200 bp, aby zapewnić lepszą wydajność amplifikacji.
4. Informacje o porównaniu sekwencji Blast, gen docelowy należy porównać w banku genów, aby zapobiec nieswoistej amplifikacji.
5. Obecność pseudogenów.Pseudogen to sekwencja DNA podobna do normalnego genu, ale traci swoją normalną funkcję.Często występuje w wielogenowej rodzinie eukariontów.Zwykle jest reprezentowany przez ψ.Jest to niefunkcjonalna kopia genomowego DNA w genomie, która jest bardzo podobna do kodującej sekwencji genu., na ogół nie są transkrybowane i nie mają jasnego znaczenia fizjologicznego.
6. Pozycja starterów względem eksonów i intronów.We wczesnych latach, kiedy rozwiązaliśmy problem zanieczyszczenia DNA, często zwracaliśmy uwagę na pozycje starterów, eksonów i intronów i ogólnie rozważaliśmy zaprojektowanie starterów w poprzek intronów, aby uniknąć amplifikacji DNA.Proszę spojrzeć na poniższy rysunek: czarny reprezentuje introny, różne błękity reprezentują eksony, różowy reprezentuje wspólne startery, a jaskrawoczerwony reprezentuje startery obejmujące introny.
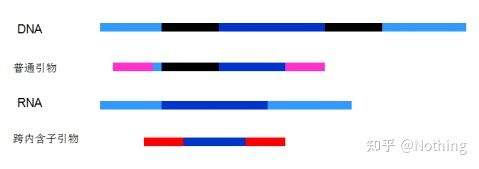
Schematyczne, nigdy prawdziwe
Wydaje się to idealnym planem, ale w rzeczywistości w większości przypadków startery trans-intronowe nie są tak magiczne, jak sobie wyobrażano, a także spowodują niespecyficzną amplifikację.Tak więc najlepszym sposobem zapobiegania zanieczyszczeniu DNA jest całkowite usunięcie DNA.
7. Przewidywanie konformacji.Korzystając ponownie z tego przykładu, użyj narzędzia online mFold, aby określić drugorzędową strukturę docelowego fragmentu w określonej temperaturze i stężeniu soli.
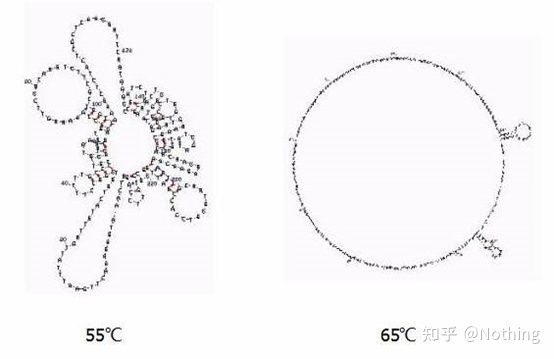
Struktura drugorzędowa RNA w różnych temperaturach
Struktura drugorzędowa to komplementarne parowanie samej matrycy, co doprowadzi do silnej konkurencji między parowaniem startera i matrycy, a szanse na związanie startera są mniejsze, co skutkuje szeregiem problemów, takich jak niska E i słaba powtarzalność.Dzięki przewidywaniu oprogramowania, jeśli nie ma problemu ze strukturą drugorzędową, byłoby świetnie.Jeśli tak, w naszym kolejnym artykule szczegółowo omówimy sposób rozwiązania tego problemu.
MIQE (6) — oligonukleotydy qPCR

W przypadku fluorescencyjnego ilościowego PCR pierwszą rzeczą, z którą zmagasz się każdego dnia, jest ekstrakcja RNA, a drugą rzeczą może być projektowanie starterów.
Przede wszystkim nadal sprawdzamy zasady projektowania podkładów zgodnie z listą kontrolną MIQE.To jest tak proste, że szumowiny mogą się śmiać, a my możemy zakończyć to jednym zdaniem: znajdź sekwencję i pozycję sondy startera oraz metodę modyfikacji.W przypadku metody oczyszczania starterów synteza starterów jest obecnie tak tania, że qPCR jest wart PAGE i wyższych metod oczyszczania, a informacje o instrumencie do syntezy nie są ważne.Wiele osób robi podkłady od dziesięcioleci i nie wie, że syntezator to ABI3900.
Jeśli chodzi o zasady projektowania podkładów, nie musisz zapamiętywać ich na pamięć, ponieważ większość programów do projektowania podkładów lub narzędzi online może rozwiązać te problemy (zalecane narzędzie online primer3.ut.ee/), a 99,999% projektowania podkładów nie jest wykonywane ręcznie.
Wystarczy sprawdzić następujące punkty po zaprojektowaniu podkładów:
1. Zaprojektuj startery blisko końca 3′: W przypadku wykorzystania starterów oligo dT do syntezy pierwszej nici cDNA, biorąc pod uwagę wydajność odwrotnej transkrypcji i integralność RNA, projektowane startery należy zaprojektować blisko końca 3′, aby poprawić wydajność amplifikacji.Użyj ilustracji, aby wyjaśnić w następujący sposób (nie ma sposobu, aby to zrozumieć):
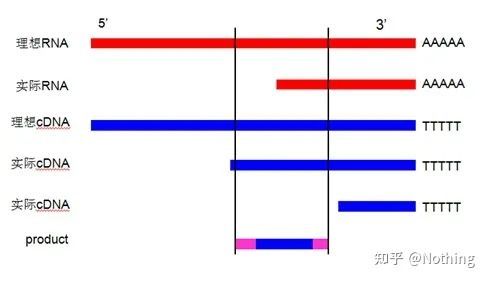
Dlaczego startery miałyby być projektowane blisko końca 3′, to nie może być prawda
2. Wartość TM: Wartość Tm wynosi 55-65°C (ponieważ aktywność egzonukleazy jest najwyższa w temperaturze 60°C), a zawartość GC wynosi 40%-60%.
3. BLAST: Aby uniknąć niespecyficznej amplifikacji genomu, należy użyć Blast do dodatkowej weryfikacji.
MIQE(7) — proces qPCR
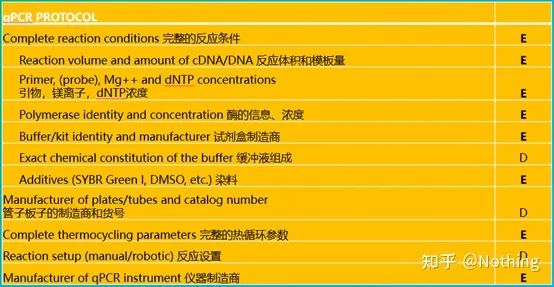
1. Zestaw qPCR
Zgodnie z wymaganiami MIQE, w artykule musimy jasno opisać kompletne warunki reakcji, w tym konfigurację systemu reakcji PCR, jaki zestaw jest używany, kto jest producentem, jak duży jest system reakcji, czy stosowana jest metoda barwnika, czy metoda sondy, ustawienia programu PCR.Doświadczeni kierowcy z pewnością stwierdzą, że o ile zestaw jest wybrany, powyższe informacje są w zasadzie zdeterminowane.
Obecnie wytwarzanie zestawów fluorescencyjnych ilościowych PCR jest technologią bardzo dojrzałą.Dopóki nie wybierzesz wyjątkowo złych producentów, prawdopodobieństwo problemów nie jest wysokie, ale nadal chcemy podzielić się z Tobą kilkoma punktami:
Enzym Taq z gorącym startem:Najważniejszą częścią PCR jest enzym Taq typu hot-start.Enzymy typu hot-start dostępne na rynku są generalnie podzielone na dwa typy, jeden to chemicznie modyfikowany enzym typu hot-start (można go sobie wyobrazić jako zatapianie w parafinie), a drugi to enzym typu hot-start do modyfikacji przeciwciał (wiązanie antygen-przeciwciało).Modyfikacja chemiczna to wczesny sposób enzymów o gorącym rozruchu.Po osiągnięciu określonej temperatury enzym uwalnia swoją aktywność.Zmodyfikowany przeciwciałem enzym gorącego startu wykorzystuje metody biologiczne do blokowania aktywności enzymu.Po osiągnięciu określonej temperatury przeciwciało zostanie zdenaturowane i zdezaktywowane jako białko, a aktywność enzymatyczna zostanie włączona.
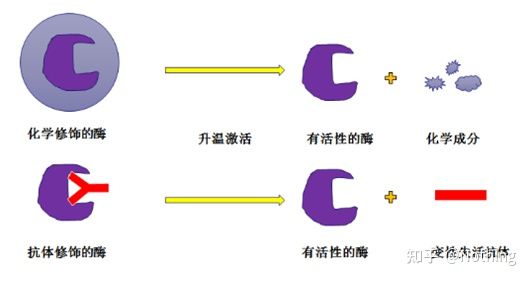
Jednak jaki jest z tego pożytek?W tym przypadku aktywność uwalniania enzymów modyfikowanych przeciwciałami jest szybsza niż enzymów modyfikowanych chemicznie, więc pod względem czułości enzymy modyfikowane przeciwciałami mają niewielką przewagę, tak że w zestawach na rynku zasadniczo nie ma enzymów modyfikowanych chemicznie.Jeśli tak, to technologia tego producenta wciąż tkwi w epoce tysiąclecia.
Stężenie jonów magnezu:Stężenie jonów magnezu jest bardzo ważne w reakcji PCR.Odpowiednie stężenie jonów magnezu może sprzyjać uwalnianiu aktywności enzymu Taq.Jeśli stężenie jest zbyt niskie, aktywność enzymu zostanie znacznie zmniejszona;jeśli stężenie jest zbyt wysokie, wzmocniona zostanie niespecyficzna amplifikacja katalizowana przez enzym.Stężenie jonów magnezu będzie również wpływać na hybrydyzację starterów, temperaturę topnienia matrycy i produktów PCR, wpływając w ten sposób na wydajność zamplifikowanych fragmentów.Stężenie jonów magnezu na ogół kontroluje się na poziomie 25 mM.Oczywiście dla dobrego zestawu stężenie jonów magnezu musi być dobrze kontrolowane.Niektórzy kupcy dodają do odczynnika środek chelatujący jony magnezu, który może osiągnąć efekt automatycznej regulacji stężenia jonów magnezu.
Stężenie barwnika fluorescencyjnego:Barwnik fluorescencyjny, którym zwykle jest SYBR Green, generuje fluorescencję głównie poprzez wiązanie się z mniejszym rowkiem dwuniciowego DNA, ponieważ wiązanie barwnika z dwuniciowym DNA jest niespecyficzne, to znaczy tak długo, jak dwuniciowy DNA jest z nim połączony, może wystąpić fluorescencja, więc dimery starterów i matryce DNA w systemie połączą się z nim, tworząc sygnał tła.
PS: Ze względu na właściwości światłoczułe produkty dostępne na rynku są zwykle pakowane w brązowe nieprzezroczyste probówki wirówkowe (jak pokazano na poniższym rysunku).Spowoduje to jednak napotkanie problemu.Podczas pobierania próbki trudno jest stwierdzić, czy ciecz jest zasysana.Pod tym względem Qingke jest rzeczywiście najbardziej przyjazne dla użytkownika (co widać na poniższym obrazku), a przezroczysta tuba jest zapakowana w nieprzezroczystą blaszaną torebkę.Następnie umieść go w blaszanym woreczku, biorąc pod uwagę wygodę unikania światła i pobierania próbek.Musisz wybrać właściwy numer produktu.TSE204 to super opłacalna egzystencja, która sprawia, że chcę sadzić trawę.

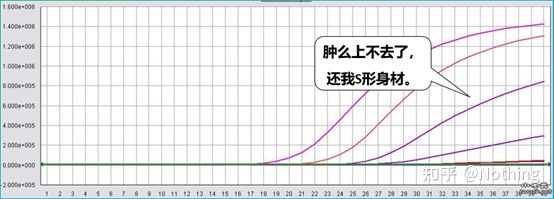
Bardzo ważne jest również stężenie barwnika fluorescencyjnego.Jeśli stężenie jest zbyt niskie, krzywa amplifikacji nie wzrośnie w późniejszym etapie i nie będzie idealna;jeśli stężenie jest zbyt wysokie, spowoduje to zakłócenia hałasu.Ponieważ fluorescencyjny ilościowy PCR zależy głównie od wartości CT, jeśli stężenie barwnika fluorescencyjnego nie jest odpowiednio wyregulowane, najniższy punkt jest lepszy niż najwyższy punkt.Oczywiście najlepsze jest odpowiednie stężenie barwnika.
ROX: Barwniki ROX są używane do korygowania błędów sygnału fluorescencji między dołkami.Niektórzy producenci przyrządów wymagają kalibracji, a inni nie.Na przykład użycie aparatu do amplifikacji PCR w czasie rzeczywistym firmy Thermo Fisher Scientific zwykle wymaga kalibracji, w tym 7300, 7500, 7500Fast, StepOnePlus itp. Opisują to ogólne instrukcje zestawu.
Foregene's qPCR Mix zawiera również barwnik ROX, który jest wygodny w użyciu w różnych modelach.
Zestaw Real Time PCR Kit-Taqman
Leczenie słabymi wiązaniami wodorowymi: Leczenie słabych wiązań wodorowych jest sprawą stosunkowo techniczną.Nikt nie czytał instrukcji wielu zestawów, ale żaden z nich nie poruszył tego tematu.W rzeczywistości jest to bardzo ważne.Kombinacja zasad zależy głównie od siły wiązań wodorowych.Silne wiązania wodorowe są normalną amplifikacją, a słabe wiązania wodorowe prowadzą do niespecyficznej amplifikacji.Jeśli nie można dobrze wyeliminować słabych wiązań wodorowych, nie można uniknąć amplifikacji niespecyficznej.W zakresie autora tylko kilka firm zauważyło ten problem.Kupując zestaw, możesz odnieść się do tego, czy rozważałeś rozwiązanie w tym zakresie dla zestawu, który chcesz wybrać.
Objętość reakcji: Częściej używany jest system 20-50 ul, a mniejsze objętości mogą powodować błędy.Ogólnie rzecz biorąc, instrukcja zestawu będzie zalecać stosowanie objętości reakcji PCR.Nie bądź mądry i używaj mniejszych ilości, aby zaoszczędzić na kosztach.cel.Ilość zalecana przez sprzedawców została faktycznie przetestowana i może się zdarzyć, że nie będą w stanie rozwiązać problemu błędów spowodowanych małymi ilościami.
2. Producent i numer artykułu płyty rurowej
Każdy zna zasadę fluorescencyjnego ilościowego PCR.Zbieranie fluorescencji odbywa się głównie przez korki do probówek PCR.Wybierając materiały eksploatacyjne do PCR, zwróć uwagę na dwa punkty: dobrą przepuszczalność światła i dopasowanie do aparatu.Ogólnie rzecz biorąc, deski i tuby mainstreamowych marek są w porządku, ale trzeba ostrożnie wybierać pod względem adaptacji, inaczej nie będziesz mógł korzystać z instrumentu.

4. Wiedza na najwyższym poziomie
MIQE (8) — walidacja qPCR
To najwyższy priorytet qPCR!Tak wielu bohaterów upadło tutaj w piasek.Oczywiście możliwe jest również, że masz szczęście i geny, które badałeś, są proste, więc płynąłeś przez lodową jaskinię z wiatrem.Informacje weryfikacyjne qPCR mają na celu sprawdzenie wiarygodności danych.Niezbędne informacje weryfikacyjne podajemy w następujący sposób:
1.Test specyficzności
Specyficzność amplifikacji genu docelowego sprawdza się, sprawdzając, czy obraz elektroforetyczny jest pojedynczym prążkiem;weryfikacja sekwencjonowania;krzywą topnienia, aby sprawdzić, czy mapa piku jest pojedyncza;weryfikacja trawienia enzymatycznego i inne metody.
Tutaj skupiamy się na tAnaliza amplifikacji nieswoistej metodą krzywych topnienia.Mówiąc ogólnie, kiedy projektujemy startery, wymagana jest wielkość fragmentu produktu w zakresie 80-200 bp, co sprawia, że temperatura topnienia produktu PCR mieści się w zakresie 80-85°C.Dlatego, jeśli występują różne piki, muszą istnieć inne niespecyficzne produkty amplifikacji;jeśli pik pojawia się poniżej 80°C, ogólnie uważa się, że jest to dimer startera;jeśli pik pojawia się powyżej 85°C, ogólnie uważa się, że jest to zanieczyszczenie DNA lub bardziej niespecyficzna amplifikacja dużych fragmentów.
Uwaga: Czasami występuje tylko jeden pik przy 80°C.W tej chwili należy przestrzegać tej koncepcji.Jest prawdopodobne, że wszystkie wyniki amplifikacji to dimery starterów.
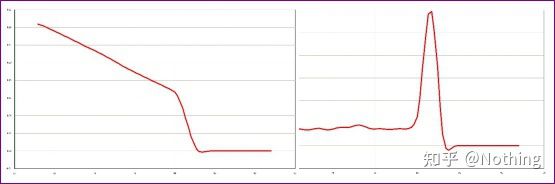
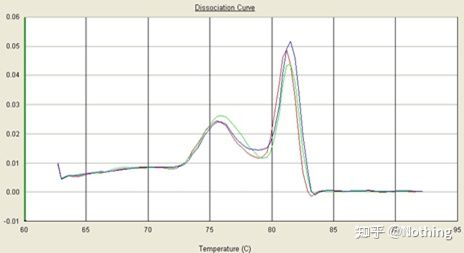
Normalna krzywa topnienia (pojedynczy pik bez niespecyficznej amplifikacji)
Problematyczna krzywa topnienia (niespecyficzne wzmocnienie fałszywych pików)
【Analiza spraw】
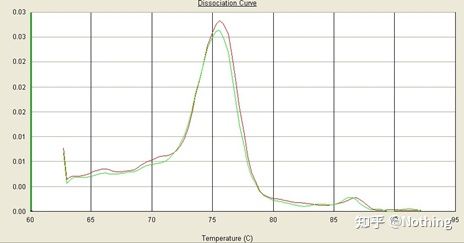
Jest główny pik, ale dimer startera jest poważny
Krzywa topnienia z pojedynczym pikiem na poniższym rysunku może łatwo oszukać twoje oczy, myśląc, że to doskonały eksperyment, ale wynik jest całkowicie błędny.W tym czasie musimy patrzeć na temperaturę topnienia.Temperatura piku wynosi poniżej 80°C, co jest całkowitym dimerem startera.
Brak docelowego fragmentu, wszystkie dimery starterów
Tutaj mój brat nie może przestać.Poniższe zdjęcie to zdjęcie zrobione telefonem komórkowym wysłane do mnie przez szumowinę.Odczynniki, których użył, to wszystkie powszechnie stosowane marki w branży.Zmienił markę z prefiksem T na inną markę z prefiksem T.Myślę, że już się domyśliłeś.Łajdak krzyknął do mnie: „Odczynnik użyty na pierwszym zdjęciu jest za dobry, a pik jest pojedynczy.Później, po użyciu zalecanego odczynnika, staje się jak na drugim obrazku, z mieszanymi pikami.Uczyniłeś mnie nieszczęśliwą.“
Rozdziel oba wykresy.Na pierwszy rzut oka jeden ma pojedynczy szczyt, a drugi podwójny.Nonsens, jeden szczyt jest oczywiście w porządku.Czy to prawda?
Gorzej niż Dou E, jeśli umieszczę dwa zdjęcia na poniższym obrazku, natychmiast zrozumiesz.W rzeczywistości łatwo nas sparaliżować tego rodzaju obrazem.Po dokładnej analizie stwierdziliśmy, że: pik na pierwszej figurze występuje w temperaturze 75°C, która jest całkowitym dimerem startera;pik na drugiej cyfrze pojawia się przy 75°C i 82°C, przynajmniej tam jest. Pojawia się produkt.
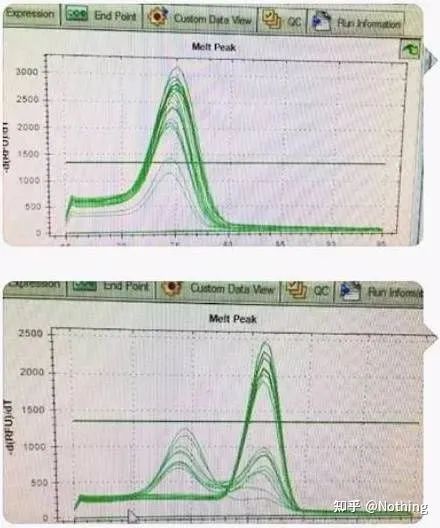
Zdjęcia opinii uczniów
Tak więc podstawowym problemem nie jest problem odczynników, ale problem projektowania starterów.Jednocześnie dowodzi to również, że niektóre duże marki nie mają jakości żelaza, a także potwierdza to, co powiedział wcześniej mój brat: To nie marka odczynników wspiera twój artykuł.To twój artykuł podtrzymał markę odczynników.Wyobraź sobie, że gdyby ten drań nie zmienił odczynników, do dziennika trafiłyby błędne dane i doszłoby do tragedii.
2. Wartość Ct próby ślepej
Nie wyjaśniaj, jeśli ślepa próba kontrolna ma wartość Ct, czy nie jest to zanieczyszczenie?Jednak nadal musisz zrozumieć, która kontrola ślepa ma wartość Ct.Jeśli jest to NTC, oznacza to obecność obcego DNA, takiego jak zanieczyszczenie odczynnikiem.Jeśli jest to NRT, oznacza to, że wyekstrahowany RNA ma zanieczyszczenie DNA.
3. Krzywa standardowa
Uwzględniając nachylenie i wzór obliczeniowy, wydajność PCR można obliczyć za pomocą wzoru.Idealny eksperyment wymaga, aby nachylenie krzywej standardowej zbliżyło się do 3,32, a R² zbliżyło się do 0,9999.
4. Liniowy zakres dynamiczny
Zakres dynamiczny reakcji jest liniowy.Zgodnie z szablonem użytym do wygenerowania krzywej wzorcowej, zakres dynamiczny powinien zawierać co najmniej 5 gradientów stężeń i zwrócić uwagę na zmianę wartości Ct przy dużych gradientach stężeń i małych gradientach stężeń.
5. Dokładność wykrywania
Zmiany w wynikach qPCR, czyli słaba powtarzalność, czyli słaba precyzja, są spowodowane wieloma czynnikami, w tym temperaturą, stężeniem i działaniem.Precyzja qPCR generalnie staje się trudniejsza do kontrolowania wraz ze spadkiem liczby kopii.W idealnej sytuacji zmienność w obrębie eksperymentu, ta techniczna zmienność powinna być odmienna od biologicznej, a repliki biologiczne mogą bezpośrednio odnosić się do różnic statystycznych w wynikach qPCR między grupami lub zabiegami.W szczególności w przypadku testów diagnostycznych należy podać najlepszą precyzję między testami (powtarzalność) w różnych ośrodkach i operatorach.
6. Skuteczność detekcji i LOD (w multipleksowym qPCR)
LOD to najniższe stężenie z 95% wykrytych próbek dodatnich.Innymi słowy, stężenie LOD zawarte w zestawie replikatów genu docelowego nie powinno przekraczać 5% nieudanych reakcji.Podczas przeprowadzania multipleksowej analizy qPCR, zwłaszcza w celu jednoczesnego wykrywania mutacji punktowych lub polimorfizmów, multipleksowa metoda qPCR musi dostarczyć dowodów na to, że dokładność wielu fragmentów docelowych nie jest zagrożona w tej samej probówce, wydajność wykrywania wielokrotnego i wykrywania w pojedynczej probówce powinna być taka sama.Zwłaszcza gdy geny docelowe o wysokim stężeniu i geny docelowe o niskim stężeniu są jednocześnie amplifikowane, należy zwrócić uwagę na ten problem.
Problemy i rozwiązaniaOgólnie rzecz biorąc, problemy często napotykane podczas debugowania qPCR koncentrują się na następujących aspektach:
·amplifikacja niespecyficzna
·Trudny wybór stężenia starterów i kłopoty z dimerami starterów
· Temperatura wyżarzania jest niedokładna
·Drugorzędowa struktura wpływa na wydajność amplifikacji
amplifikacja niespecyficzna
amplifikacja niespecyficznazdarza się, ogólnie uważa się, czy konstrukcja podkładu nie jest odpowiednia, ale jeśli nie spieszy ci się ze zmianą podkładów, możesz najpierw wypróbować następujące metody (zasada jest również załączona):
·Podwyższyć temperaturę wyżarzania – postarać się, aby słabe wiązania wodorowe nie były w stanie utrzymać się;
·Krótszy czas wyżarzania i wydłużania – zmniejsza ryzyko powstania słabych wiązań wodorowych;
·Zmniejsz stężenie starterów – zmniejsz ryzyko wiązania zbędnych starterów i regionów niedocelowych;
Niska wydajność wzmocnienia
Sytuacja odwrotna do amplifikacji niespecyficznej – niska wydajność amplifikacji, a środki do radzenia sobie z niską wydajnością amplifikacji są wręcz odwrotne:
· Przedłużyć czas wyżarzania i wydłużenia;
·Zmiana na trójetapową reakcję PCR i obniżenie temperatury hybrydyzacji;
·Zwiększyć stężenie podkładu;
Ps: Wielu absolwentów urodzonych w latach 90. nie chce uczyć się, jak debugować eksperymenty i ma nadzieję, że zestaw może całkowicie rozwiązać problem (jeśli chcesz iść do firmy odczynnikowej, aby prowadzić badania i rozwój po ukończeniu studiów), w rzeczywistości producenci odczynników również myślą w ten sposób, mam nadzieję, że to głupiec Można go użyć, gdy go zdobędziesz, więc producenci odczynników poświęcili wiele wysiłku, aby rozwiązać problem niespecyficznej amplifikacji, w tym wprowadzenie słabych współczynników absorpcji wiązania H.Aby łatwo rozwiązać problem, głupcy wciąż muszą przeczytać wstęp firmy odczynnikowej, aby zobaczyć, czy istnieje czynnik, który absorbuje słabe wiązania wodorowe.
Trudny dobór stężenia starterów i kłopoty ze starterami-dimerami
Metoda 1: Ogólnie rzecz biorąc, instrukcje zestawu do qPCR zawierają zalecane systemy i zalecane stężenia starterów.
Metoda 2: Debugowanie poprzez ustawienie gradientu stężenia startera.Poniższe zdjęcie zostało skradzione z firmy w celu zilustrowania.Poniższy rysunek przedstawia ilościowe wyniki fluorescencji wykonane z trzema gradientami stężeń starterów (100 nM, 250 nM, 500 nM) i czterema gradientami stężeń matrycy (0,1 ng, 1 ng, 10 ng, 100 ng).Wartość Ct wyników eksperymentalnych jest wykreślana w następujący sposób:
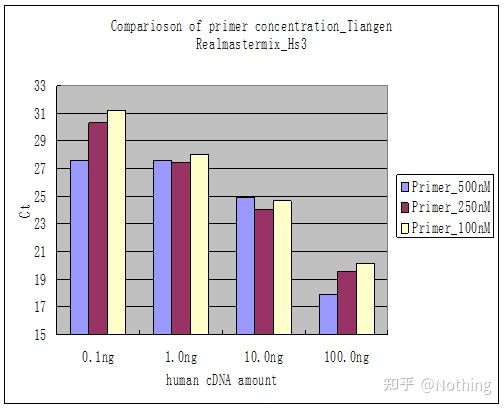
Wybór stężenia startera Połącz każde stężenie startera w linię w następujący sposób:
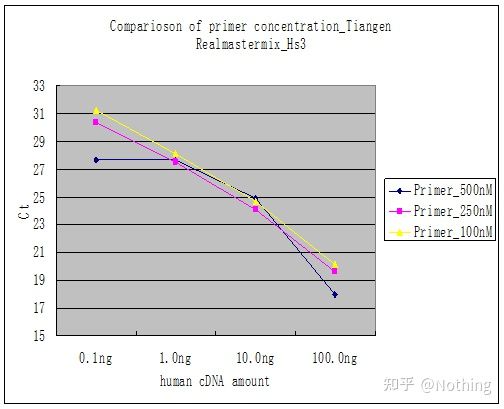
Wybór stężenia startera jest oczywisty, liniowa zależność stężenia startera 100nM i 250nM jest lepsza, a liniowa zależność stężenia startera 500nM jest stosunkowo słaba.W 100nM i 250nM wartość Ct 250nM jest stosunkowo mała, więc optymalne stężenie startera wynosi 250nM.Ogólnie na krzywej topnienia można zobaczyć silne startery-dimery.Co jeśli zaprojektowane startery nie mogą uniknąć starterów-dimerów?
Metoda 3: Zmniejsz ilość starterów i zwiększ temperaturę wyżarzania (nie trzeba wyjaśniać).
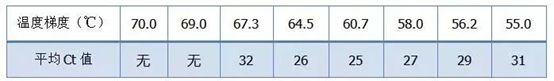
Empiryczna wartość temperatury wyżarzania wynosi 60°C.Jeśli nie jesteś pewien, jak wybrać bardziej odpowiednią temperaturę wyżarzania?Odpowiedź jest taka sama jak przy wyborze stężenia startera –test gradientu.Zrób zdjęcie z firmy Bio-rad dla zobrazowania problemu.W celu amplifikacji określonego fragmentu docelowego ustaw osiem gradientów temperatury, każdy z trzema powtórzeniami, a uzyskana krzywa amplifikacji wygląda następująco:
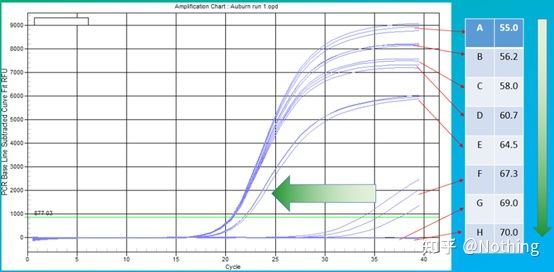
Wybór temperatury wyżarzania:
·70°C, 69°C — Zasadniczo starterów nie można łączyć, więc nie ma amplifikacji.
·67,3°C – Na początku występuje niewielka amplifikacja, a wartość Ct jest stosunkowo duża.
·64,5°C —— Wartość Ct spada.
·W 60,7°C, 58,0°C, 56,2°C i 55,0°C wartości Ct zasadniczo były stabilne, ale końcowe wartości fluorescencji były różne.
Jak wybrać?Zasada: Pierwszą zasadą jest wyższa wartość Ct.Dla tej samej wartości Ct wybierz wyższą temperaturę hybrydyzacji, aby uniknąć dimeryzacji i niespecyficznej amplifikacji.Chociaż wartość fluorescencji jest wyższa w temperaturze 55°C, mogą w niej występować dimery lub niespecyficzna amplifikacja.
Ale jeśli jesteś tak bystry jak ty, na pewno pomyślisz: Logicznie rzecz biorąc, jeśli reakcja PCR jest bardzo specyficzna, o ile stężenie startera przekracza minimalne wymagania, wysokie i niskie punkty nie powinny mieć wpływu, podobnie jak barwniki fluorescencyjne i dNTP.Rzeczywiście, dopóki temperatura wyżarzania jest odpowiednio zoptymalizowana, wpływ stężenia startera na wartość Ct będzie naturalnie zminimalizowany.
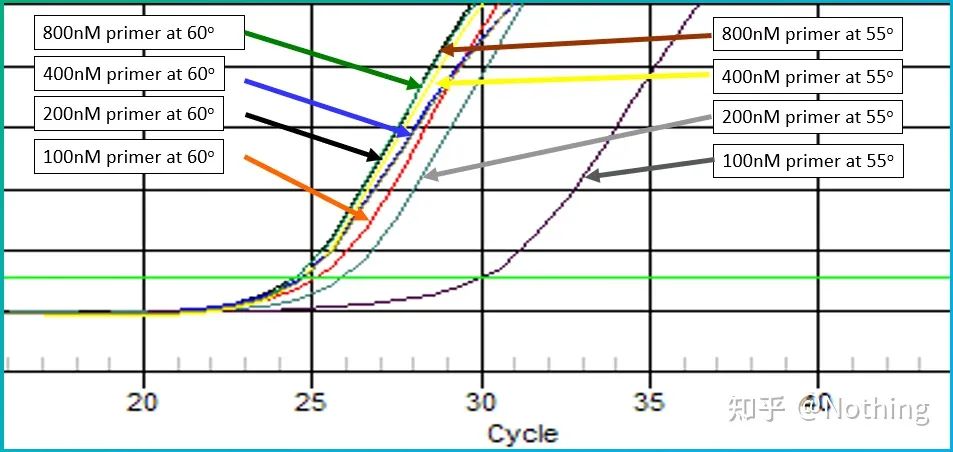
Temperatura wyżarzania jest odpowiednio zoptymalizowana, a wpływ stężenia startera na CT zostanie zminimalizowany
Struktura drugorzędowa wpływa na efektywność amplifikacji
Zróbmy zdjęcie z Bio-radu dla zilustrowania problemu.Projektuje również gradient temperatury w celu amplifikacji genu o strukturze drugorzędowej.
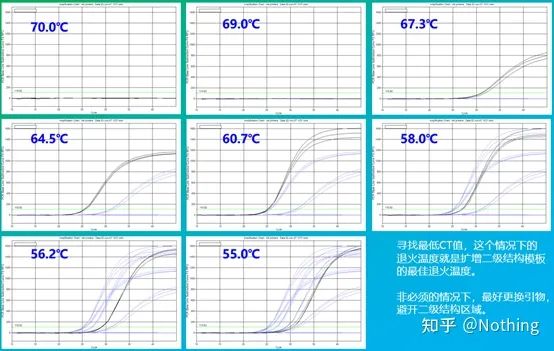
Pojawia się struktura drugorzędowa
Można zauważyć, że wraz ze spadkiem gradientu temperatury zaczynają pojawiać się produkty i wartość Ct przesuwa się do przodu, osiągając minimalną wartość przy 60,7°C, a następnie wraz ze spadkiem gradientu temperatury wartość Ct rośnie.I odwrotnie, wraz ze wzrostem temperatury struktura drugorzędowa otwiera się i zwiększa się wydajność amplifikacji.Po osiągnięciu określonej temperatury zwiększenie temperatury nie może poprawić wydajności amplifikacji.Ponieważ starterów nie można w tej chwili stabilnie połączyć.Dlatego,poszukaj temperatury o najniższej wartości Ct, która jest najlepszą temperaturą do amplifikacji szablonu struktury drugorzędowej!Oczywiście sprytni głupcy muszą wiedzieć, że jeśli nie jest to konieczne, najlepiej zmienić startery i uniknąć regionu struktury drugorzędowej.
5. Poziom aplikacji
MIQE — analiza danych

Analiza danych jest przeprowadzana głównie za pomocą instrumentu fluorescencyjnej ilościowej reakcji PCR.W poprzednim artykule wykonano wiele prac związanych z analizą danych, takich jak ślepa próba kontrolna, co zostało wyjaśnione w projekcie eksperymentu.Wyjaśniono wewnętrzne geny referencyjne, liczby powtórzeń itp., tutaj wyjaśniamy głównie zastosowanie qPCR.
qPCR jest szeroko stosowany, a weryfikacja eksperymentalna i diagnostyka kwasów nukleinowych to najczęściej stosowane scenariusze.
bezwzględna kwantyfikacja
Log (stężenie początkowe) ma liniową zależność od liczby cykli.Krzywą wzorcową można wykreślić ze wzorca o znanej początkowej liczbie kopii, to znaczy można uzyskać liniową zależność reakcji amplifikacji.Na podstawie wartości Ct próbki można obliczyć stężenie w próbce.Liczba szablonów do uwzględnienia.
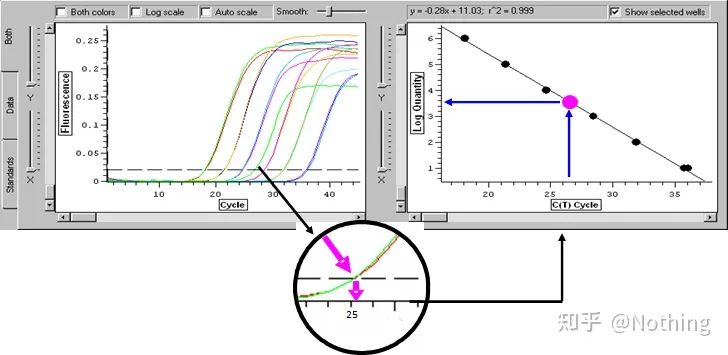
Bezwzględna ilościowa metoda obliczeniowa
Kwantyfikacja bezwzględna musi opierać się na krzywej wzorcowej.Aby wykonać krzywą standardową, wymagany jest standard.Zwykle standardem jest plazmid otrzymany przez sklonowanie docelowego genu.Dlaczego jest plazmidem?Ponieważ okrągły plazmidowy DNA jest najbardziej stabilny.Rozcieńczyć standardowy produkt w 5 do 6 gradientach zgodnie ze stosunkiem podwojenia (10-krotne rozcieńczenie) i zwrócić uwagę na jednorodność podczas rozcieńczania.Niech wartość Ct spadnie między 15-30.
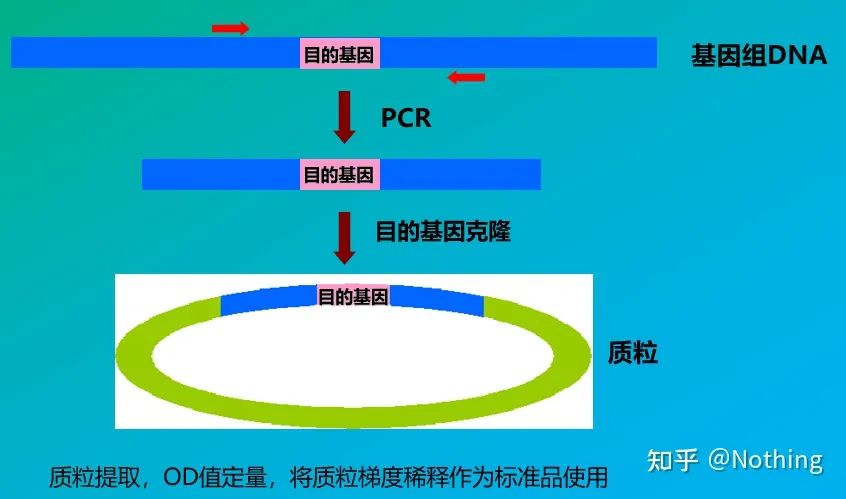
Przygotowanie standardowe
Równocześnie badaną próbkę również należy odpowiednio rozcieńczyć (pamiętać o współczynniku rozcieńczenia), a wartość Ct również powinna mieścić się w przedziale 15-30.Standardowy produkt + próbka do badania są umieszczane razem na maszynie.Po serii sporządzono krzywą wzorcową z substancją wzorcową, a próbki przeznaczone do badania umieszczono na krzywej wzorcowej w celu obliczenia stężenia.
Oznaczanie ilościowe HBV wirusa zapalenia wątroby typu B jest typowym oznaczaniem bezwzględnym, które pozwala obliczyć liczbę kopii wirusa w 1 ml krwi.
Obliczanie liczby kopii
Stężenie badanej próbki (ng/ul) = OD260 × 50 ug/ml × współczynnik rozcieńczenia
Masa cząsteczkowa próbki = liczba zasad × 324
Liczba kopii badanej próbki (kopie/ul) = stężenie badanej próbki / masa cząsteczkowa próbki × 6 × 1014

Metoda obliczania liczby kopii
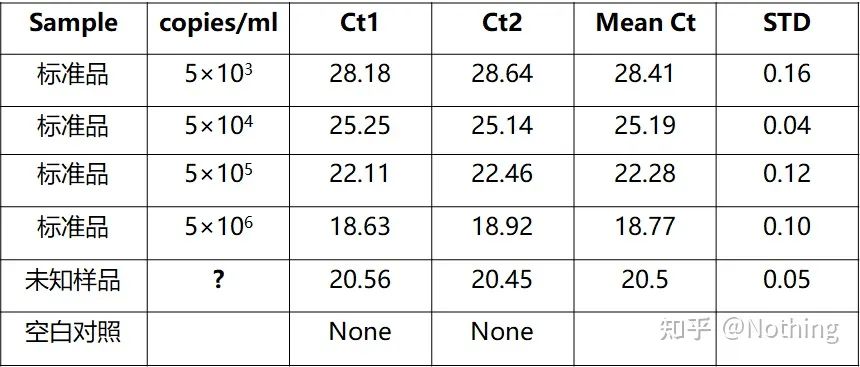
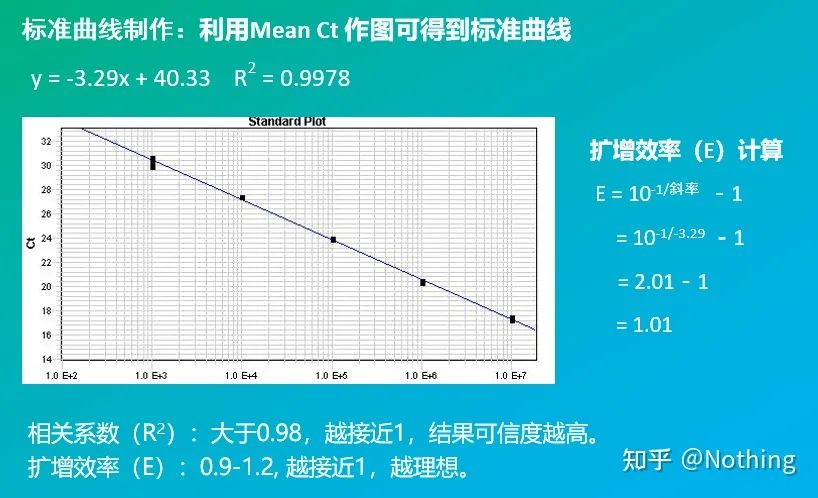

Powyżej przedstawiono metodę obliczeniową służącą do określania ilości.Jest to problem matematyczny, który można rozwiązać po ukończeniu gimnazjum, a problemy matematyczne rozwiązują na ogół komputery.Jeśli nie rozumiesz, możesz przyjść, aby się porozumieć.
względna kwantyfikacja
Kwantyfikacja względna jest stosowana głównie w badaniach naukowych.Ile wirusów znajduje się w 1 ml krwi i jest to wirus DNA, jest to zdarzenie względnie deterministyczne: ilość krwi można określić, a wirus DNA jest względnie stabilny.Jednak trudno jest nam porównać liczbę kopii transkrypcyjnych określonego genu w liściu, ponieważ trudno jest określić rozmiar, wagę i delikatność liścia, trudno jest określić ilość wyekstrahowanego RNA, trudno jest również określić wydajność odwrotnej transkrypcji, to znaczy każdy krok może sprawić, że dane eksperymentalne będą zawierały błędy i nie będą mogły zostać wykorzystane.
Dlatego względna kwantyfikacja musi wprowadzić element:wewnętrzny gen odniesienia.
Innymi słowy, względna ocena ilościowa jest w rzeczywistości porównaniem genu docelowego z wewnętrznym genem referencyjnym.W porównaniu z tą samą tkanką i tą samą komórką wpływ wielkości próbki, ilości ekstrakcji RNA, wydajności odwrotnej transkrypcji i wydajności PCR jest stosunkowo niewielki.Ze względu na mały rozmiar próbki, zarówno wewnętrzne geny referencyjne, jak i geny docelowe były stosunkowo zmniejszone.Dlatego już wcześniej kładliśmy nacisk na jednolitość i stabilność.
Wewnętrzne geny referencyjne są na ogółgeny sprzątania(geny utrzymujące dom), które odnoszą się do klasy genów, które są stabilnie wyrażane we wszystkich komórkach, a ich produkty są niezbędne do utrzymania podstawowych czynności życiowych komórek.
Nie myl tego pojęcia.Geny porządkowe to terminy funkcji biologicznych, podczas gdy wewnętrzne geny odniesienia to eksperymentalne terminy techniczne.Geny porządkowe muszą przejść walidację, zanim będą mogły zostać wybrane jako wewnętrzne geny referencyjne.
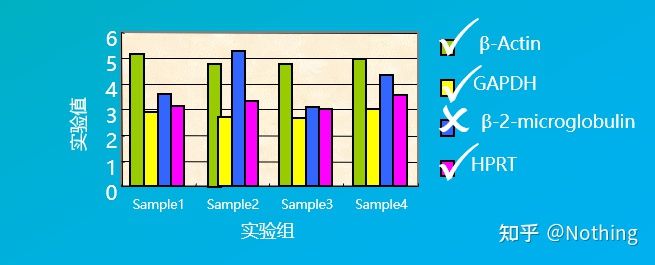
Na przykład wybraliśmy kilka genów porządkowych na poniższym rysunku, aby przetestować ich poziomy ekspresji w różnych komórkach tkankowych i stwierdziliśmy, że poziomy ekspresji β-2-mikroglobuliny były zupełnie różne od poziomów pozostałych trzech genów, więc nie mogły być używane jako wewnętrzne geny referencyjne.
Po zrozumieniu funkcji korekcyjnej wewnętrznego genu referencyjnego wyprowadza się dwa algorytmy dzięki wprowadzeniu wewnętrznego genu referencyjnego.
·metoda podwójnej krzywej standardowej
·2 – Metoda △△Ct (metoda porównywania wartości CT)
Jeśli jesteś zainteresowany badaniem gatunków i funkcji genów, proszę porzuć badania nad algorytmami i używaj bezpośrednio wzorów lub bezpośrednio używaj maszyn;jeśli jesteś prostym facetem z matematyki i inżynierii, nie krępuj się.
metoda podwójnej krzywej standardowej
Oceń ilościowo gen docelowy i gen metabolizmu podstawowego próbki kontrolnej i próbki, która ma być testowana, za pomocą krzywej standardowej, a następnie oblicz względną wartość zgodnie ze wzorem obliczeniowym, która jest względnym poziomem ekspresji.
Zalety: prosta analiza, stosunkowo prosta optymalizacja eksperymentalna
Wada: Dla każdego genu każda runda eksperymentów musi tworzyć krzywą standardową
Zastosowanie: Jedna z dwóch najczęściej stosowanych i uznanych względnych metod ilościowych w badaniu regulacji ekspresji genów
Formuła jest następująca:

Przykłady są następujące:
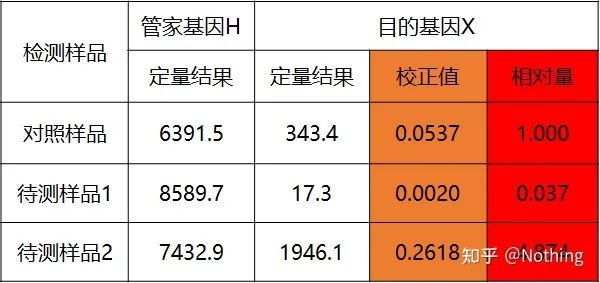
Oblicz względną kwotę na podstawie wyniku ilościowego
2 – Metoda △△Ct (metoda porównywania wartości CT)

Zalety: Nie ma potrzeby wykonywania krzywej wzorcowej
Wady: Przyjmuje się, że skuteczność wzmocnienia jest bliska 100%;odchylenie standardowe wynosi < 5% i przyjmuje się, że krzywa standardowa i wydajność pomiędzy poszczególnymi amplifikacjami są spójne;optymalizacja warunków doświadczalnych jest bardziej skomplikowana.
Zastosowanie: Jedna z dwóch najczęściej stosowanych i uznanych względnych metod ilościowych w badaniu regulacji ekspresji genów
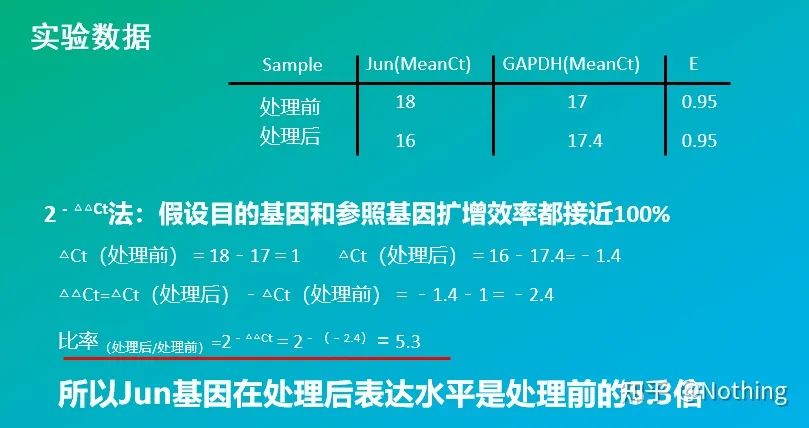
Oczywiście wydajność amplifikacji jest zwykle niemożliwa do osiągnięcia w sposób doskonały 1. Metoda korekcji: Jeśli wiemy, że gen docelowy i gen referencyjny mają taką samą wydajność amplifikacji, ale wydajność amplifikacji nie jest równa 1, to 2-△△Ct można skorygować jako: (1+E )-△△Ct, na przykład, jeśli wydajność amplifikacji wynosi 0,95, wówczas wzór obliczeniowy można skorygować do 1,95- △△Ct
Jak dotąd treść dotycząca fluorescencyjnego ilościowego PCR dobiegła końca.
Czas postu: 06-04-2023



