



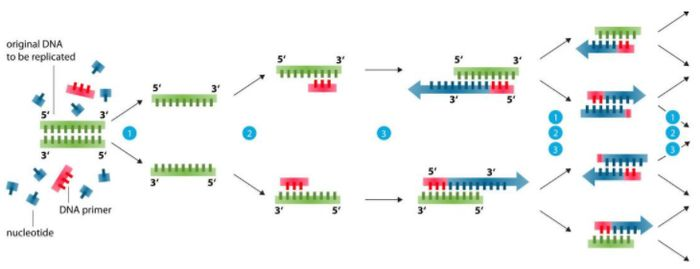
Podstawa projektowania podkładu (99% problemów można rozwiązać)
1. Długość startera: Podręcznik wymaga 15-30 pz, zwykle około 20 pz.Rzeczywisty stan powinien wynosić 18-24 bp, aby zapewnić specyficzność, ale im dłuższy, tym lepszy, zbyt długi starter również zmniejszy specyficzność i zmniejszy wydajność.
2. Rozpiętość amplifikacji startera: odpowiednia jest 200-500 bp, a fragment można rozszerzyć do 10 kb w określonych warunkach.
3. Baza podkładowa: Zawartość G+C powinna wynosić 40-60%, zbyt mała amplifikacja G+C nie jest dobra, zbyt duża ilość G+C łatwo powoduje pojawienie się niespecyficznych prążków.ATGC najlepiej rozprowadza się losowo, unikając skupisk więcej niż 5 nukleotydów purynowych lub pirymidynowych.Multi-gc dla sekwencji końca 5' i sekwencji pośrednich w celu zwiększenia stabilności, unikaj bogatej GC na końcu 3', bez GC dla ostatnich 3 zasad lub bez GC dla 3 z ostatnich 5 zasad.
4. Unikaj struktury drugorzędowej w starterach i unikaj komplementacji między dwoma starterami, zwłaszcza komplementacji na końcu 3', w przeciwnym razie powstanie dimer startera i zostaną wygenerowane niespecyficzne amplifikowane prążki.
5. Zasady na 3'-końcu starterów, zwłaszcza ostatnie i przedostatnie zasady, powinny być ściśle sparowane, aby uniknąć niepowodzenia PCR z powodu niesparowanych zasad końcowych.
6. Startery mają lub mogą być dodane z odpowiednimi miejscami cięcia, a amplifikowana sekwencja docelowa powinna korzystnie mieć odpowiednie miejsca cięcia, co jest bardzo korzystne dla analizy cięcia lub klonowania molekularnego.
7. Specyficzność starterów: startery nie powinny wykazywać oczywistej homologii z innymi sekwencjami w bazie danych sekwencji kwasów nukleinowych.
8. Naucz się korzystać z oprogramowania: PP5, Oligo6, DNAstar, Vector NTI, primer3 (ten projekt online działa najlepiej).
Powyższa treść może rozwiązać co najmniej 99% problemów projektowych podkładów.
Kontroluj szczegóły projektu podkładu
1. Długość podkładu
Ogólna długość startera wynosi 18~30 zasad.Ogólnie rzecz biorąc, najważniejszym czynnikiem determinującym temperaturę wygrzewania podkładu jest długość podkładu.Zwykle wybiera się temperaturę wyżarzania podkładu (wartość Tm -5 ℃), a niektóre bezpośrednio wykorzystują wartość Tm.Poniższe wzory można z grubsza obliczyć temperaturę wygrzewania podkładów.
Gdy długość startera jest mniejsza niż 20 bp: [4(G+C)+2(A+T)]-5℃
Gdy długość startera jest większa niż 20 bp: 62,3 ℃ + 0,41 ℃ (%GC) -500 / długość -5 ℃
Ponadto wiele programów może być również używanych do obliczania temperatury wyżarzania, zasada obliczeń będzie inna, więc czasami obliczona wartość może mieć niewielką lukę.Aby zoptymalizować reakcje PCR, stosuje się najkrótsze startery, które zapewniają temperaturę hybrydyzacji nie niższą niż 54 ℃, co zapewnia najlepszą wydajność i specyficzność.
Ogólnie rzecz biorąc, specyficzność startera wzrasta czterokrotnie dla każdego dodatkowego nukleotydu, tak że minimalna długość startera dla większości zastosowań wynosi 18 nukleotydów.Górna granica długości startera nie jest bardzo istotna, związana głównie z wydajnością reakcji.Ze względu na entropię, im dłuższy starter, tym mniejsza szybkość, z jaką łączy się on z docelowym DNA, tworząc stabilną dwuniciową matrycę, z którą wiąże się polimeraza DNA.
Używając oprogramowania do projektowania starterów, długość starterów można z kolei określić na podstawie wartości TM, szczególnie w przypadku starterów do ilościowego PCR fluorescencji, TM = 60 ℃ lub mniej więcej powinno być kontrolowane.
2. Zawartość GC
Generalnie zawartość G+C w sekwencjach starterów wynosi 40%~60%, a zawartość GC i wartość Tm pary starterów powinny być skoordynowane.Jeśli starter ma poważną tendencję do GC lub AT, odpowiednią ilość ogona A, T lub G i C można dodać do 5'-końca startera.
3. Temperatura wyżarzania
Temperatura wyżarzania powinna być o 5 ℃ niższa niż temperatura rozkuwania.Jeśli liczba zasad starterów jest niewielka, temperaturę przyłączania można odpowiednio zwiększyć, co może zwiększyć specyficzność PCR.Jeśli liczba zasad jest duża, temperaturę wyżarzania można odpowiednio obniżyć.Różnica temperatur hybrydyzacji między parą starterów wynosząca 4 ℃ ~ 6 ℃ nie wpłynie na wydajność PCR, ale idealnie temperatura hybrydyzacji pary starterów jest taka sama i może wahać się między 55 ℃ ~ 75 ℃.
4. Unikaj obszaru struktury drugorzędowej matrycy do amplifikacji
Najlepiej jest unikać regionu struktury drugorzędowej matrycy podczas wybierania zamplifikowanego fragmentu.Stabilną strukturę drugorzędową docelowego fragmentu można przewidzieć i oszacować za pomocą odpowiedniego oprogramowania komputerowego, co jest pomocne przy wyborze matrycy.Wyniki eksperymentów pokazują, że ekspansja często kończy się niepowodzeniem, gdy energia swobodna (△G) obszaru, który ma zostać rozszerzony, jest mniejsza niż 58,6 kJ/mol.
5. Niezgodność z docelowym DNA
Gdy zamplifikowana docelowa sekwencja DNA jest duża, starter może wiązać się z wieloma częściami docelowego DNA, co skutkuje pojawieniem się wielu prążków w wyniku.Tym razem konieczne jest skorzystanie z testowania oprogramowania BLAST, strona internetowa:http://www.ncbi.nlm.nih.gov/BLAST/.Wybierz Wyrównaj dwie sekwencje (bl2seq).
Wklejanie sekwencji starterów do strefy 1 i docelowych sekwencji DNA do strefy 2 jest wymienne, a BLAST oblicza komplementarność, antysensowność i inne możliwości, więc użytkownicy nie muszą zauważać, czy oba łańcuchy są łańcuchami sensownymi.Możesz również wprowadzić numer GI, jeśli znasz numer GI sekwencji w bazie danych, dzięki czemu nie musisz wklejać dużej części sekwencji.Na koniec kliknij opcję Align at 3, aby sprawdzić, czy starter ma wiele miejsc homologicznych w docelowym DNA.
6. Końcówka podkładu
Koniec 3 startera to miejsce, w którym zaczyna się wydłużanie, dlatego ważne jest, aby zapobiegać rozpoczynaniu się niedopasowań w tym miejscu.Koniec 3' nie powinien znajdować się dalej niż 3 kolejne G lub C, ponieważ spowoduje to błędne wyzwolenie startera w regionie sekwencji wzbogacania G+C.Koniec 3' nie może tworzyć żadnej struktury drugorzędowej, z wyjątkiem specjalnych reakcji PCR (AS-PCR), koniec 3' startera nie może być niedopasowany.Na przykład, jeśli region kodujący jest amplifikowany, koniec 3' startera nie powinien być zakończony w trzeciej pozycji kodonu, ponieważ trzecia pozycja kodonu jest podatna na degenerację, co wpłynie na specyficzność i wydajność amplifikacji.Używając starterów do aneksji, należy zapoznać się z tabelą wykorzystania kodonów, zwrócić uwagę na preferencje biologiczne, nie używać starterów do aneksji na końcu 3' i stosować wyższe stężenie starterów (1 uM-3 uM).
7. Struktura drugorzędowa starterów
Same startery nie powinny mieć komplementarnych sekwencji, w przeciwnym razie same startery zwiną się w struktury szpilki do włosów, a ta drugorzędowa struktura wpłynie na wiązanie starterów i matryc z powodu zawady sterycznej.Jeśli stosuje się sztuczną ocenę, ciągłe komplementarne zasady samych starterów nie powinny być większe niż 3 bp.Nie powinno być komplementarności między dwoma starterami, zwłaszcza należy unikać komplementarnego nakładania się końca 3', aby zapobiec tworzeniu się dimerów starterów.Na ogół nie powinno być więcej niż 4 kolejnych zasad homologii lub komplementarności między parą starterów.
8. Dodaj znaczniki lub loci
Koniec 5' ma niewielki wpływ na specyficzność amplifikacji i dlatego może być modyfikowany bez wpływu na specyficzność amplifikacji.Modyfikacja końca startera 5' obejmowała: dodanie miejsca restrykcyjnego enzymu;Znakowana biotyna, fluorescencja, digoksyna, Eu3+ itp. Wprowadzenie sekwencji DNA wiążących białka;Wprowadzanie miejsc mutacji, wstawianie i brakujące sekwencje mutacji oraz wprowadzanie sekwencji promotora itp. Dodatkowe zasady w mniejszym lub większym stopniu wpłyną na wydajność amplifikacji i zwiększą szansę na utworzenie dimeru startera, ale w następnym kroku należy poczynić pewne ustępstwa.Dodatkowe sekwencje, które nie występują w sekwencji docelowej, takie jak miejsca restrykcyjne i sekwencje promotora, można dodać do końca 5' startera bez wpływu na specyficzność.Sekwencje te nie są uwzględniane przy obliczaniu wartości Tm starterów, ale należy je przetestować pod kątem komplementarności i wewnętrznej struktury drugorzędowej.
9. Subklony
W większości przypadków PCR to tylko wstępne klonowanie, a następnie musimy subklonować docelowy fragment do różnych wektorów, więc musimy zaprojektować dodatkowe zasady do następnej operacji w etapie PCR.
Poniżej podsumowano niektóre sekwencje przeznaczone do subklonowania.
Dodano miejsce restrykcyjne dla endonukleazy restrykcyjnej
Dodawanie miejsc restrykcyjnych dla enzymów jest najczęściej stosowaną metodą subklonowania produktów PCR.Ogólnie rzecz biorąc, miejsce rozszczepienia to sześć zasad, oprócz 5 'końca miejsca rozszczepienia należy dodać 2 ~ 3 zasady ochronne.Jednak liczba zasad ochronnych wymaganych przez różne enzymy jest różna.Na przykład SalⅠ nie wymaga podstawek ochronnych, EcoRⅤ wymaga 1 podstawek ochronnych, NotⅠ wymaga 2 podstawek ochronnych, a Hind Ⅲ wymaga 3 podstawek ochronnych.
LIC dodaje ogon
Pełna nazwa LIC to Ligation-Independent Cloning, metoda klonowania wymyślona przez firmę Navogen specjalnie dla tej części wektora pET.Nośnik pET przygotowany metodą LIC ma niekomplementarne lepkie końce pojedynczej nici o długości 12-15 zasad, które uzupełniają odpowiednie lepkie końce docelowego fragmentu wstawki.Dla celów amplifikacji sekwencja startera 5' wstawionego fragmentu powinna uzupełniać wektor LIC.Aktywność ekstraktu 3′→5′ polimerazy DNA T4 może po krótkim czasie utworzyć lepki koniec pojedynczej nici na wstawionym fragmencie.Ponieważ produkt może powstać jedynie w wyniku wzajemnego wyżarzania przygotowanego fragmentu wstawki i wektora, metoda ta jest bardzo szybka i wydajna i polega na klonowaniu ukierunkowanym.
Skierowany klon TA dodaje ogon
Klonowanie TA nie było w stanie skierować fragmentu do wektora, więc później Invitrogen wprowadził wektor, który mógł celować w klonowanie, który zawierał cztery wyróżniające się zasady GTGGS na jednym końcu.Dlatego przy projektowaniu starterów do PCR należy odpowiednio dodawać sekwencje komplementarne, aby fragmenty mogły być „orientowane”.
Jeśli masz mało czasu, możesz spróbować bezpośredniej syntezy, łącząc gen z wektorem, co u muskularystów nazywamy syntezą genu ET.
D. Metoda klonowania w fuzji
Nie wymaga ligazy, nie wymaga długiej reakcji.Dopóki sekwencje na obu końcach linearyzowanego wektora zostaną wprowadzone do projektowania starterów, a następnie produkt PCR i linearyzowany wektor zostaną dodane do roztworu enzymu do infuzji zawierającego BSA i umieszczone w temperaturze pokojowej na pół godziny, można przeprowadzić transformację.Ta metoda jest szczególnie odpowiednia do konwersji dużych objętości.
10. Scal podkład
Czasami znane są tylko ograniczone informacje o sekwencji dotyczące projektu startera.Na przykład, jeśli znana jest tylko sekwencja aminokwasowa, można zaprojektować starter łączący.Starter łączący to mieszanina różnych sekwencji reprezentujących wszystkie różne możliwości zasad, które kodują pojedynczy aminokwas.Aby zwiększyć specyficzność, możesz skorzystać z tabeli wykorzystania kodonów, aby zmniejszyć aneksję zgodnie z podstawowymi preferencjami różnych organizmów.Hipoksantynę można łączyć ze wszystkimi zasadami, aby obniżyć temperaturę wyżarzania podkładu.Nie używać dołączonych zasad na końcu 3' startera, ponieważ hybrydyzacja ostatnich 3 zasad na końcu 3' jest wystarczająca do zainicjowania PCR w niewłaściwym miejscu.Stosowane są wyższe stężenia starterów (1 μM do 3 μM), ponieważ startery w wielu mieszaninach do aneksji nie są specyficzne dla docelowej matrycy.
Surowce PCRkontrola
1. Ilość podkładu
Stężenie każdego startera wynosi 0,1 ~ 1 umol lub 10 ~ 100 pmol.Lepiej jest uzyskać wymagany rezultat przy użyciu najmniejszej ilości podkładu.Wysokie stężenie startera spowoduje niedopasowanie i niespecyficzną amplifikację oraz zwiększy szansę na utworzenie dimerów między starterami.
2. Stężenie startera
Stężenie starterów wpływa na specyficzność.Optymalne stężenie startera wynosi na ogół od 0,1 do 0,5 μM.Wyższe stężenia starterów prowadzą do amplifikacji nieswoistych produktów.
3. Temperatura wyżarzania podkładu
Innym ważnym parametrem podkładów jest temperatura topnienia (Tm).Jest to temperatura, w której 50% starterów i sekwencji komplementarnych jest reprezentowanych jako dwuniciowe cząsteczki DNA.Tm jest wymagana do ustawienia temperatury hybrydyzacji PCR.Idealnie, temperatura hybrydyzacji jest wystarczająco niska, aby zapewnić skuteczne hybrydyzację starterów z sekwencją docelową, ale wystarczająco wysoka, aby zmniejszyć wiązanie niespecyficzne.Rozsądna temperatura wyżarzania od 55 ℃ do 70 ℃.Temperatura wyżarzania jest na ogół o 5 ℃ niższa niż Tm podkładu.
Istnieje kilka wzorów ustalania Tm, które różnią się znacznie w zależności od użytego wzoru i sekwencji starterów.Ponieważ większość wzorów podaje szacunkową wartość Tm, wszystkie temperatury wyżarzania są tylko punktem wyjścia.Specyficzność można poprawić, analizując kilka reakcji, które stopniowo podnoszą temperaturę hybrydyzacji.Rozpocznij poniżej szacowanej Tm-5 ℃ i stopniowo zwiększaj temperaturę wyżarzania w krokach co 2 ℃.Wyższa temperatura wyżarzania zmniejszy tworzenie się dimerów starterów i niespecyficznych produktów.Aby uzyskać najlepsze wyniki, dwa startery powinny mieć przybliżone wartości Tm.Jeśli różnica Tm par starterów jest większa niż 5 ℃, startery wykażą znaczny falstart dzięki zastosowaniu niższej temperatury hybrydyzacji w cyklu.Jeśli dwa startery Tm są różne, ustaw temperaturę wyżarzania o 5 ℃ niższą niż najniższa Tm.Alternatywnie, w celu zwiększenia specyficzności, można najpierw przeprowadzić pięć cykli w temperaturach hybrydyzacji zaprojektowanych dla wyższej Tm, a następnie pozostałe cykle w temperaturach hybrydyzacji zaprojektowanych dla niższej Tm.Pozwala to na uzyskanie częściowej kopii docelowego szablonu w ściśle określonych warunkach.
4. Czystość i stabilność podkładu
Standardowa czystość niestandardowych starterów jest odpowiednia dla większości zastosowań PCR.Usunięcie grup benzoilowych i izobutylowych przez odsalanie jest minimalne i dlatego nie zakłóca reakcji PCR.Niektóre zastosowania wymagają oczyszczania w celu usunięcia sekwencji niepełnej długości w procesie syntezy.Te skrócone sekwencje występują, ponieważ wydajność chemii syntezy DNA nie wynosi 100%.Jest to proces okrężny, który wykorzystuje powtarzające się reakcje chemiczne, ponieważ każda zasada jest dodawana w celu wytworzenia DNA od 3 ′ do 5 ′.Możesz zawieść w każdym cyklu.Dłuższe startery, zwłaszcza te dłuższe niż 50 zasad, mają dużą część skróconych sekwencji i mogą wymagać oczyszczania.
Na wydajność starterów ma wpływ wydajność chemii syntezy i metoda oczyszczania.Firmy biofarmaceutyczne, takie jak Cytology i Shengong, wszystkie stosują minimalną jednostkę OD, aby zapewnić całkowitą wydajność oligonukleozydu.Niestandardowe podkłady są dostarczane w postaci suchego proszku.Najlepiej ponownie rozpuścić startery w TE tak, aby końcowe stężenie wynosiło 100μM.TE jest lepsze niż woda dejonizowana, ponieważ pH wody jest często kwaśne i powoduje hydrolizę oligonukleozydów.
Stabilność podkładów zależy od warunków przechowywania.Suchy proszek i rozpuszczone podkłady należy przechowywać w temperaturze -20℃.Podkłady rozpuszczone w TE w stężeniach większych niż 10 μM można stabilnie przechowywać w temperaturze -20 ℃ przez 6 miesięcy, ale można je przechowywać tylko w temperaturze pokojowej (15 ℃ do 30 ℃) krócej niż 1 tydzień.Suche podkłady proszkowe można przechowywać w temperaturze -20 C przez co najmniej 1 rok iw temperaturze pokojowej (15 C do 30 C) do 2 miesięcy.
5. Enzymy i ich stężenia
Obecnie stosowana polimeraza DNA Taq jest w zasadzie enzymem inżynierii genowej syntetyzowanym przez bakterie z grupy coli.Ilość enzymu potrzebna do katalizowania typowej reakcji PCR wynosi około 2,5 U (odnosi się do całkowitej objętości reakcji 100 ul).Jeśli stężenie jest zbyt wysokie, może to prowadzić do niespecyficznej amplifikacji;jeśli stężenie jest zbyt niskie, ilość syntetycznego produktu zostanie zmniejszona.
6. Jakość i stężenie dNTP
Jakość dNTP jest ściśle związana ze stężeniem i wydajnością amplifikacji PCR.Proszek dNTP jest granulowany, a jego zmienność traci swoją aktywność biologiczną, jeśli jest niewłaściwie przechowywany.Roztwór dNTP jest kwaśny i powinien być stosowany w wysokim stężeniu, z 1M roztworem NaOH lub 1M roztworem buforowym Tris.HCL, aby dostosować jego pH do 7,0 ~ 7,5, niewielka ilość opakowania jednostkowego, przechowywanie w stanie zamrożonym w temperaturze -20 ℃.Wielokrotne zamrażanie i rozmrażanie spowoduje degradację dNTP.W reakcji PCR dNTP powinno wynosić 50 ~ 200umol/L.Szczególnie należy zwrócić uwagę na to, aby stężenie czterech DNTPS było równe (preparat równomolowy).Jeśli stężenie któregokolwiek z nich różni się od pozostałych (wyższe lub niższe), spowoduje to niedopasowanie.Zbyt niskie stężenie zmniejszy wydajność produktów PCR.dNTP może łączyć się z Mg2+ i zmniejszać stężenie wolnego Mg2+.
7. Matrycowy (gen docelowy) kwas nukleinowy
Ilość i stopień oczyszczenia matrycowego kwasu nukleinowego jest jednym z kluczowych ogniw decydujących o powodzeniu lub niepowodzeniu PCR.Tradycyjne metody oczyszczania DNA zwykle wykorzystują SDS i proteazę K do trawienia i usuwania próbek.Główne funkcje SDS to: rozpuszczanie lipidów i białek na błonie komórkowej, niszcząc w ten sposób błonę komórkową poprzez rozpuszczanie białek błonowych i dysocjowanie białek jądrowych w komórce, SDS może również łączyć się z białkami i wytrącać;Proteaza K może hydrolizować i trawić białka, zwłaszcza histony związane z DNA, a następnie używać rozpuszczalnika organicznego, fenolu i chloroformu, do ekstrakcji białek i innych składników komórek, a także używać etanolu lub alkoholu izopropylowego do wytrącania kwasu nukleinowego.Wyekstrahowany kwas nukleinowy może być użyty jako matryca do reakcji PCR.W przypadku ogólnych próbek do wykrywania klinicznego można zastosować szybką i prostą metodę rozpuszczania komórek, lizowania patogenów, trawienia i usuwania białek z chromosomów w celu uwolnienia genów docelowych oraz bezpośredniego zastosowania do amplifikacji PCR.Ekstrakcja matrycy RNA zwykle wykorzystuje metodę izotiocyjanianu guanidyny lub proteazy K, aby zapobiec degradacji RNA przez RNazę.
8. Stężenie Mg2+
Mg2+ ma istotny wpływ na specyficzność i wydajność amplifikacji PCR.W ogólnej reakcji PCR, gdy stężenie różnych dNTP wynosi 200umol/L, odpowiednie stężenie Mg2+ wynosi 1,5 ~ 2,0 mmol/L.Stężenie Mg2+ jest zbyt wysokie, specyficzność reakcji spada, następuje amplifikacja niespecyficzna, zbyt niskie stężenie obniży aktywność polimerazy DNA Taq, powodując redukcję produktów reakcji.
Jony magnezu wpływają na kilka aspektów PCR, takich jak aktywność polimerazy DNA, która wpływa na wydajność;Innym przykładem jest wyżarzanie starterów, które wpływa na specyficzność.dNTP i matryca wiążą się z jonem magnezu, zmniejszając ilość wolnego jonu magnezu wymaganego do aktywności enzymu.Optymalne stężenie jonów magnezu jest różne dla różnych par starterów i matryc, ale typowe stężenie początkowe PCR z 200 μM dNTP wynosi 1,5 mM (uwaga: do ilościowego PCR w czasie rzeczywistym należy użyć 3 do 5 mM roztworu jonów magnezu z sondą fluorescencyjną).Wyższe stężenia wolnych jonów magnezu zwiększają wydajność, ale także zwiększają amplifikację niespecyficzną i zmniejszają wierność.Aby określić optymalne stężenie, przeprowadzono miareczkowanie jonów magnezu w krokach co 0,5 mM od 1 mM do 3 mM.Aby zmniejszyć zależność od optymalizacji jonów magnezu, można zastosować polimerazę Platinum Taq DNA.Platynowa polimeraza DNA Taq jest zdolna do utrzymania funkcji w szerszym zakresie stężeń jonów magnezu niż polimeraza DNA Taq i dlatego wymaga mniejszej optymalizacji.
9. Dodatki promujące PCR
Optymalizacja temperatury wyżarzania, projektu startera i stężenia jonów magnezu jest wystarczająca do wysoce specyficznej amplifikacji większości matryc;jednak niektóre szablony, w tym te o wysokiej zawartości GC, wymagają dodatkowych środków.Dodatki wpływające na temperaturę topnienia DNA to kolejny sposób na poprawę specyficzności produktu i wydajności.Aby uzyskać najlepsze wyniki, wymagana jest pełna denaturacja szablonu.
Ponadto struktura drugorzędowa zapobiega wiązaniu starterów i wydłużaniu enzymów.
Dodatki PCR, w tym formamid, DMSO, gliceryna, betaina i PCRx Enhancer Solution, wzmacniają amplifikację.Ich możliwym mechanizmem jest obniżenie temperatury topnienia, wspomagając w ten sposób hybrydyzację starterów i wspomagając wydłużanie polimerazy DNA przez region struktury drugorzędowej.Rozwiązanie PCRx ma jeszcze inne zalety.W przypadku stosowania z polimerazą DNA Platinum Taq i polimerazą DNA Platinum Pfx wymagana jest minimalna optymalizacja jonów magnezu.W ten sposób technika platynowa jest połączona z dodatkiem w celu zwiększenia specyficzności przy jednoczesnym zmniejszeniu zależności od trzeciego podejścia, optymalizacji jonów magnezu.Aby uzyskać najlepsze wyniki, należy zoptymalizować stężenie dodatków, zwłaszcza DMSO, formamidu i glicerolu, które hamują polimerazę Taq DNA.

10. Gorący start
Hot start PCR jest jedną z najważniejszych metod poprawy specyficzności PCR, oprócz dobrego zaprojektowania startera.Chociaż optymalna temperatura wydłużania polimerazy Taq DNA wynosi 72℃, polimeraza pozostaje aktywna w temperaturze pokojowej.Zatem produkty niespecyficzne powstają, gdy temperatura przetrzymywania jest niższa niż temperatura hybrydyzacji podczas przygotowania reakcji PCR i na początku cyklu termicznego.Po utworzeniu te niespecyficzne produkty są skutecznie amplifikowane.PCR z gorącym startem jest szczególnie skuteczna, gdy miejsca użyte do zaprojektowania starterów są ograniczone przez lokalizację elementów genetycznych, takich jak mutacje ukierunkowane, klonowanie ekspresyjne lub konstrukcja i manipulacja elementami genetycznymi używanymi do inżynierii DNA.
Powszechną metodą ograniczania aktywności polimerazy DNA Taq jest przygotowanie roztworu do reakcji PCR na lodzie i umieszczenie go w ogrzanym aparacie do PCR.Metoda ta jest prosta i niedroga, ale nie dopełnia działania enzymu, a zatem nie eliminuje całkowicie amplifikacji produktów niespecyficznych.
Termiczne priming opóźnia syntezę DNA poprzez hamowanie istotnego składnika, aż aparat PCR osiągnie temperaturę denaturacji.Większość ręcznych metod inicjacji termicznej, w tym opóźnione dodawanie polimerazy DNA Taq, jest kłopotliwa, zwłaszcza w zastosowaniach o dużej przepustowości.Inne metody gruntowania termicznego wykorzystują osłonę woskową do zamknięcia istotnego składnika, w tym jonów magnezu lub enzymów, lub do fizycznej izolacji składników reaktywnych, takich jak matryce i bufory.Podczas cyklu termicznego różne składniki są uwalniane i mieszane ze sobą w miarę topienia się wosku.Podobnie jak ręczna metoda gorącego rozruchu, metoda osłony woskowej jest kłopotliwa i podatna na zanieczyszczenia i nie nadaje się do zastosowań o dużej przepustowości.
Platynowa polimeraza DNA jest wygodna i wydajna w automatycznym gorącym starcie PCR.Platynowa polimeraza Taq DNA składa się z rekombinowanej polimerazy Taq DNA połączonej z przeciwciałem monoklonalnym przeciwko polimerazie Taq DNA.Przeciwciała są formułowane przez PCR w celu hamowania aktywności enzymu podczas długotrwałego utrzymywania temperatury.Polimeraza Taq DNA została uwolniona do reakcji podczas izolacji 94 ℃ etapu denaturacji, przywracając pełną aktywność polimerazy.W przeciwieństwie do chemicznie modyfikowanej polimerazy Taq DNA do inicjacji termicznej, enzym Platinum nie wymaga przedłużonej izolacji w temperaturze 94℃ (10 do 15 minut), aby aktywować polimerazę.W przypadku polimerazy PlatinumTaq DNA 90% aktywności polimerazy Taq DNA zostało przywrócone po 2 minutach w temperaturze 94℃.

11. Nest-PCR
Kolejne rundy amplifikacji przy użyciu zagnieżdżonych starterów mogą poprawić specyficzność i czułość.Pierwsza runda to standardowa amplifikacja od 15 do 20 cykli.Małą frakcję początkowego produktu amplifikacji rozcieńczono 100 do 1000 razy i dodano do drugiej rundy amplifikacji przez 15 do 20 cykli.Alternatywnie, początkowy zamplifikowany produkt może być sortowany przez oczyszczanie na żelu.W drugiej rundzie amplifikacji stosuje się zagnieżdżony starter, który może wiązać się z sekwencją docelową wewnątrz pierwszego startera.Zastosowanie zagnieżdżonego PCR zmniejsza możliwość amplifikacji wielu miejsc docelowych, ponieważ istnieje niewiele sekwencji docelowych komplementarnych do obu zestawów starterów.Ta sama całkowita liczba cykli (30 do 40) z tymi samymi starterami amplifikowała miejsca niespecyficzne.Nested PCR zwiększa czułość ograniczonych sekwencji docelowych (np. rzadkich mrnas) i poprawia specyficzność trudnych PCRS (np. 5' RACE).
12. Opadający PCR
Descending PCR poprawia specyficzność dzięki zastosowaniu ścisłych warunków hybrydyzacji przez kilka pierwszych cykli PCR.Cykl rozpoczyna się w temperaturze wyżarzania o około 5 ℃ wyższej niż szacowana Tm, następnie każdy cykl jest zmniejszany o 1 ℃ do 2 ℃, aż temperatura wyżarzania spadnie poniżej Tm 5 ℃.Tylko docelowy szablon o najwyższej homologii zostanie zamplifikowany.Produkty te rozszerzają się w kolejnych cyklach, wypierając amplifikowane produkty niespecyficzne.Malejący PCR jest przydatny w metodach, w których stopień homologii między starterem a docelową matrycą nie jest znany, takich jak pobieranie odcisków palców DNA AFLP.
Powiązane zestawy PCR
Bohater PCR 2×TMSystem Mix ma wyższą tolerancję na inhibitory PCR niż zwykły system PCR Mix i może z łatwością poradzić sobie z amplifikacją PCR różnych złożonych matryc.Unikalny system reakcji i wysoka wydajność Taq Hero sprawiają, że reakcja PCR ma wyższą wydajność amplifikacji, specyficzność i czułość.
Wyższa wydajność wzmocnienia
Ma aktywność polimerazy DNA 5'→3' i aktywność egzonukleazy 5'→3', bez aktywności egzonukleazy 3'→5'.
Zestaw Real Time PCR Easyᵀᴹ-SYBR Green I
Specyficzny — zoptymalizowany bufor i enzym Taq typu hot-start mogą zapobiegać amplifikacji niespecyficznej i tworzeniu się dimeru startera
Wysoka czułość — może wykrywać niskie kopie szablonu
Zestaw wykorzystuje unikalny odczynnik Foregene do odwrotnej transkrypcji i polimerazę Foregene HotStar Taq DNA w połączeniu z unikalnym systemem reakcji, aby skutecznie poprawić wydajność amplifikacji i specyficzność reakcji.
Czas postu: maj-09-2023



