



RT-qPCR wywodzi się ze zwykłej technologii PCR.Dodaje fluorescencyjne chemikalia (barwniki fluorescencyjne lub sondy fluorescencyjne) do tradycyjnego systemu reakcji PCR i wykrywa proces hybrydyzacji i wydłużania PCR w czasie rzeczywistym zgodnie z ich różnymi mechanizmami luminescencyjnymi.Zmiany sygnału fluorescencyjnego w pożywce są wykorzystywane do obliczenia wielkości zmiany produktu w każdym cyklu PCR.Obecnie najpopularniejszymi metodami są metoda barwnika fluorescencyjnego oraz metoda sondy.
Metoda barwnika fluorescencyjnego:
Niektóre barwniki fluorescencyjne, takie jak SYBR Green Ⅰ, PicoGreen, BEBO itp., same nie emitują światła, ale emitują fluorescencję po związaniu się z mniejszym rowkiem dsDNA.Dlatego na początku reakcji PCR maszyna nie może wykryć sygnału fluorescencyjnego.Gdy reakcja przechodzi do etapu wygrzewania-wydłużania (metoda dwuetapowa) lub wydłużania (metoda trójetapowa), podwójne nici są w tym czasie otwierane, a nowa polimeraza DNA Podczas syntezy nici cząsteczki fluorescencyjne łączą się w mniejszym rowku dsDNA i emitują fluorescencję.Wraz ze wzrostem liczby cykli PCR coraz więcej barwników łączy się z dsDNA, a sygnał fluorescencyjny jest stale wzmacniany.Weźmy za przykład SYBR Green Ⅰ.
Metoda sondy:
Sonda Taqman jest najczęściej stosowaną sondą do hydrolizy.Na końcu 5' sondy znajduje się grupa fluorescencyjna, zazwyczaj FAM.Sama sonda jest sekwencją komplementarną do genu docelowego.Na końcu 3' fluoroforu znajduje się grupa gasząca fluorescencję.Zgodnie z zasadą rezonansowego transferu energii fluorescencji (rezonansowy transfer energii Förstera, FRET), gdy reporterowa grupa fluorescencyjna (cząsteczka donorowa fluorescencyjna) i wygaszająca grupa fluorescencyjna (cząsteczka fluorescencyjna akceptorowa) Gdy widma wzbudzenia nakładają się, a odległość jest bardzo mała (7-10nm), wzbudzenie cząsteczki donorowej może indukować fluorescencję cząsteczki akceptorowej, podczas gdy autofluorescencja jest osłabiona.Dlatego na początku reakcji PCR, gdy sonda jest wolna i nienaruszona w układzie, reporterowa grupa fluorescencyjna nie będzie emitować fluorescencji.Podczas hybrydyzacji starter i sonda wiążą się z matrycą.Podczas fazy wydłużania polimeraza w sposób ciągły syntetyzuje nowe łańcuchy.Polimeraza DNA ma aktywność egzonukleazy 5′-3′.Po dotarciu do sondy polimeraza DNA zhydrolizuje sondę z matrycy, oddzieli reporterową grupę fluorescencyjną od wygaszającej grupy fluorescencyjnej i uwolni sygnał fluorescencyjny.Ponieważ istnieje relacja jeden do jednego między sondą a szablonem, metoda sondy przewyższa metodę barwnika pod względem dokładności i czułości testu.
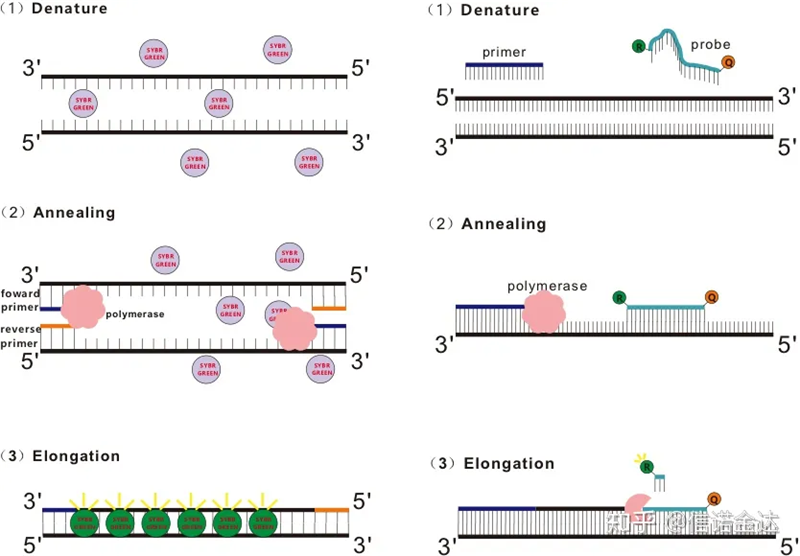
Ryc. 1 Zasada qRT-PCR
Projekt podkładu
Zasady:
Startery powinny być zaprojektowane w konserwatywnym regionie szeregu kwasów nukleinowych i wykazywać swoistość.
Najlepiej użyć sekwencji cDNA, dopuszczalna jest również sekwencja mRNA.Jeśli nie, znajdź projekt regionu cds sekwencji DNA.
Długość fluorescencyjnego produktu ilościowego wynosi 80-150 bp, najdłuższy to 300 bp, długość startera wynosi na ogół między 17-25 zasad, a różnica między starterami w górę iw dół nie powinna być zbyt duża.
Zawartość G+C wynosi od 40% do 60%, a 45-55% jest najlepsza.
Wartość TM wynosi od 58 do 62 stopni.
Staraj się unikać dimerów starterów i samodimerów (nie pojawiają się więcej niż 4 pary kolejnych komplementarnych zasad) struktura szpilki do włosów, jeśli to nieuniknione, aby ΔG<4.5kJ/mol* Jeśli nie możesz upewnić się, że gDNA zostało usunięte podczas odwrotnej transkrypcji Oczyść, najlepiej zaprojektować startery intronu *3′ koniec nie może być modyfikowany i aby uniknąć regionów bogatych w AT, GC, unikaj ciągłej struktury T/C, A/G (2-3 ) podkłady i nie-
specyficzna Homologia zamplifikowanej heterogenicznie sekwencji jest korzystnie mniejsza niż 70% lub ma homologię 8 komplementarnych zasad.
Baza danych:
Wyszukiwanie CottonFGD według słów kluczowych
Projekt podkładu:
Projekt startera IDT-qPCR
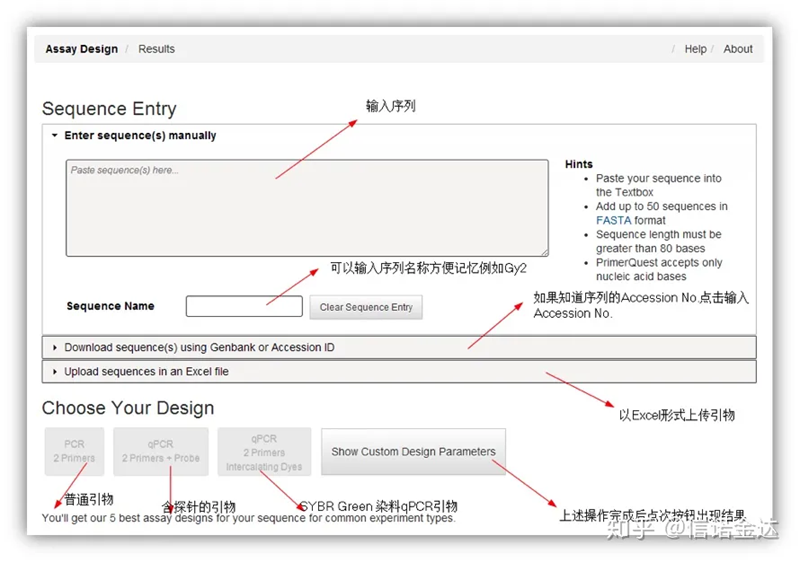
Ryc. 2 Strona internetowego narzędzia do projektowania podkładów IDT
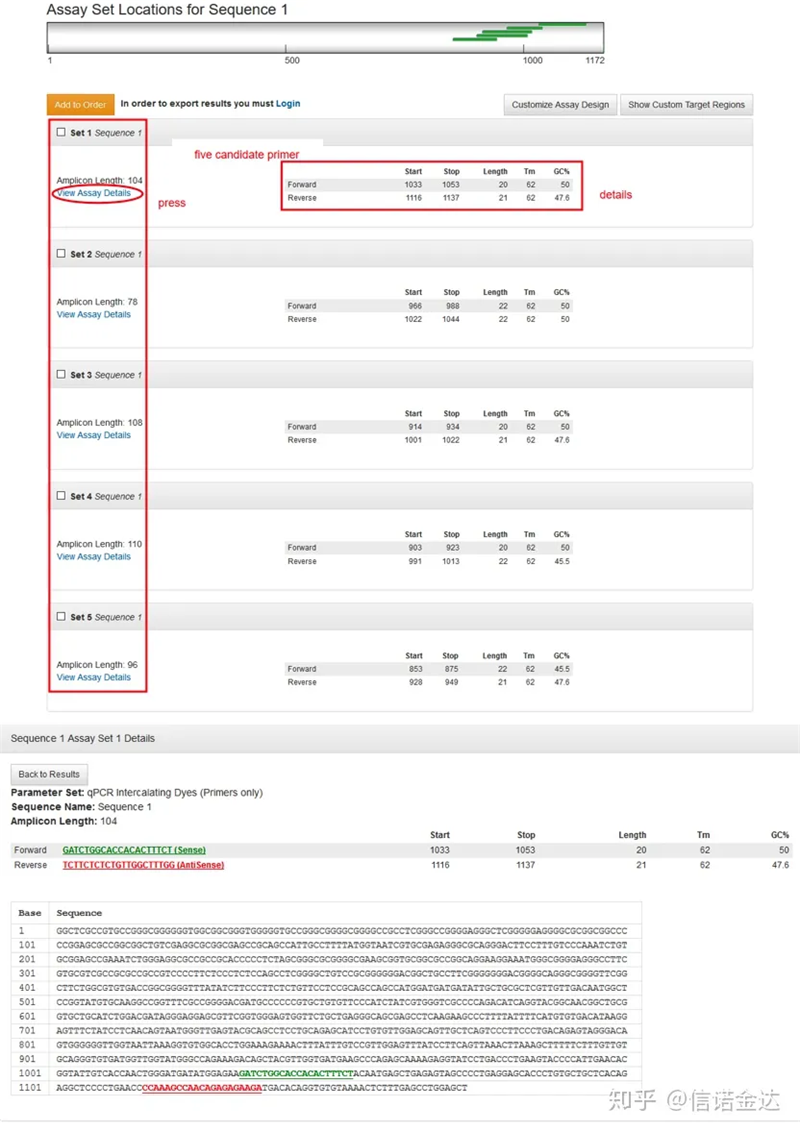
Wyświetlenie strony wyników na rys. 3
Projektowanie starterów lncRNA:
lncRNA:te same kroki co mRNA.
miRNA:Zasada metody stem-loop: Ponieważ wszystkie miRNA są krótkimi sekwencjami o długości około 23 nt, nie można przeprowadzić bezpośredniego wykrywania PCR, dlatego stosuje się narzędzie do sekwencji stem-loop.Sekwencja pnia-pętli to jednoniciowy DNA o długości około 50 nt, który sam może tworzyć strukturę spinki do włosów.3 „Koniec można zaprojektować jako sekwencję komplementarną do częściowego fragmentu miRNA, wtedy docelowy miRNA można połączyć z sekwencją łodyga-pętla podczas odwrotnej transkrypcji, a całkowita długość może osiągnąć 70 pz, co jest zgodne z długością zamplifikowanego produktu określoną metodą qPCR.Projekt startera ogonowego miRNA.
Detekcja specyficzna dla amplifikacji:
Internetowa baza danych wybuchów: wybuch CottonFGD według podobieństwa sekwencji
Lokalny wybuch: patrz użycie Blast + do lokalnego wybuchu, Linux i Macos mogą bezpośrednio ustanowić lokalną bazę danych, system Win10 można również zrobić po zainstalowaniu Ubuntu bash.Utwórz lokalną bazę danych i lokalny wybuch;otwórz bash ubuntu na win10.
Uwaga: Bawełna wyżynna i bawełna z wysp morskich to uprawy tetraploidalne, więc wynikiem wybuchu często będą dwa lub więcej dopasowań.W przeszłości użycie płyt CD NAU jako bazy danych do przeprowadzenia blastu prawdopodobnie pozwoliłoby znaleźć dwa homologiczne geny z zaledwie kilkoma różnicami SNP.Zwykle dwóch homologicznych genów nie można rozdzielić na podstawie projektu startera, więc traktuje się je jako takie same.Jeśli istnieje oczywisty indel, starter jest zwykle projektowany na indelu, ale może to prowadzić do drugorzędowej struktury startera. Energia swobodna wzrasta, co prowadzi do zmniejszenia wydajności amplifikacji, ale jest to nieuniknione.
Wykrywanie struktury drugorzędowej startera:
Kroki:otwórz oligo 7 → wprowadź sekwencję szablonu → zamknij podokno → zapisz → zlokalizuj starter na szablonie, naciśnij ctrl+D, aby ustawić długość startera → przeanalizuj różne struktury drugorzędowe, takie jak ciało samodimeryzujące, heterodimer, szpilka do włosów, niedopasowanie itp. Ostatnie dwa zdjęcia na rysunku 4 to wyniki testów starterów.Wynik przedniego podkładu jest dobry, nie ma oczywistej struktury dimeru i spinki do włosów, nie ma ciągłych komplementarnych zasad, a bezwzględna wartość energii swobodnej jest mniejsza niż 4,5, podczas gdy podkład tylny wykazuje ciągłość. 6 zasad jest komplementarnych, a energia swobodna wynosi 8,8;ponadto na końcu 3 'pojawia się poważniejszy dimer i pojawia się dimer składający się z 4 kolejnych zasad.Chociaż energia swobodna nie jest wysoka, dimer 3' Ch1 może poważnie wpływać na specyficzność amplifikacji i wydajność amplifikacji.Ponadto konieczne jest sprawdzenie spinek do włosów, heterodimerów i niedopasowań.
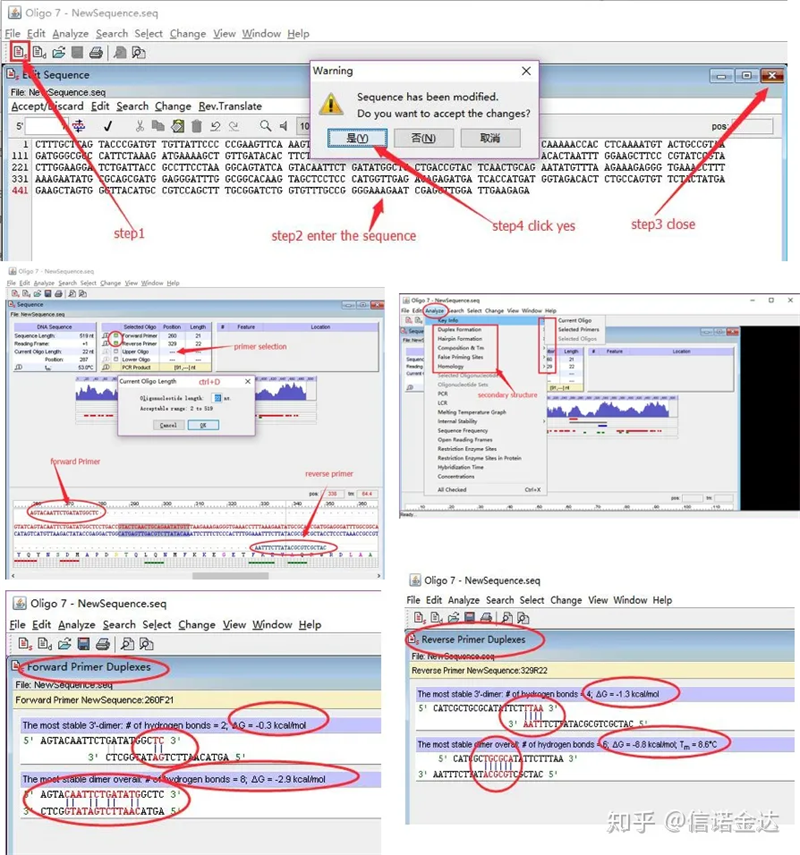
Fig. 3 Wyniki wykrywania oligo7
Wykrywanie wydajności amplifikacji:
Wydajność amplifikacji reakcji PCR poważnie wpływa na wyniki PCR.Również w qRT-PCR wydajność amplifikacji jest szczególnie ważna dla wyników ilościowych.Usuń inne substancje, maszyny i protokoły z buforu reakcyjnego.Jakość starterów ma również duży wpływ na wydajność amplifikacji qRT-PCR.Aby zapewnić dokładność wyników, zarówno względna ocena ilościowa fluorescencji, jak i bezwzględna ocena ilościowa fluorescencji muszą wykryć wydajność amplifikacji starterów.Uznaje się, że efektywna wydajność amplifikacji qRT-PCR wynosi od 85% do 115%.Istnieją dwie metody:
1. Metoda krzywej standardowej:
A.Zmieszaj cDNA
B.Rozcieńczenie gradientowe
c.qPCR
D.Równanie regresji liniowej do obliczania wydajności amplifikacji
2. LinRegPCR
LinRegPCR to program do analizy danych RT-PCR w czasie rzeczywistym, zwanych także ilościowymi danymi PCR (qPCR), opartymi na chemii SYBR Green lub podobnej.Program wykorzystuje dane nieskorygowane względem linii podstawowej, dokonuje korekty linii bazowej dla każdej próbki oddzielnie, określa okno liniowości, a następnie wykorzystuje analizę regresji liniowej w celu dopasowania linii prostej przez zestaw danych PCR.Z nachylenia tej linii obliczana jest wydajność PCR dla każdej pojedynczej próbki.Średnia wydajność PCR na amplikon i wartość Ct na próbkę są wykorzystywane do obliczenia wyjściowego stężenia na próbkę, wyrażonego w dowolnych jednostkach fluorescencji.Wprowadzanie i wyprowadzanie danych odbywa się za pomocą arkusza kalkulacyjnego Excel.Tylko próbka
wymagane jest mieszanie, bez gradientu
wymagane są kroki:(Weźmy Bole CFX96 jako przykład, niezupełnie Machine z wyraźnym ABI)
eksperyment:jest to standardowy eksperyment qPCR.
Wyjście danych qPCR:LinRegPCR rozpoznaje dwie formy plików wyjściowych: RDML lub kwantyfikacja Wynik amplifikacji.W rzeczywistości jest to wartość wykrywania numeru cyklu i sygnału fluorescencji w czasie rzeczywistym przez maszynę, a wzmocnienie uzyskuje się poprzez analizę wartości zmiany fluorescencji wydajności segmentu liniowego.
Wybór danych: Teoretycznie wartość RDML powinna być użyteczna.Szacuje się, że problem mojego komputera polega na tym, że oprogramowanie nie rozpoznaje RDML, więc mam wartość wyjściową programu Excel jako oryginalne dane.Zaleca się najpierw przeprowadzić zgrubną selekcję danych, np. nieudane dodanie próbek itp. Punkty można usunąć w danych wyjściowych (oczywiście nie można ich usunąć, LinRegPCR zignoruje te punkty w późniejszym etapie)
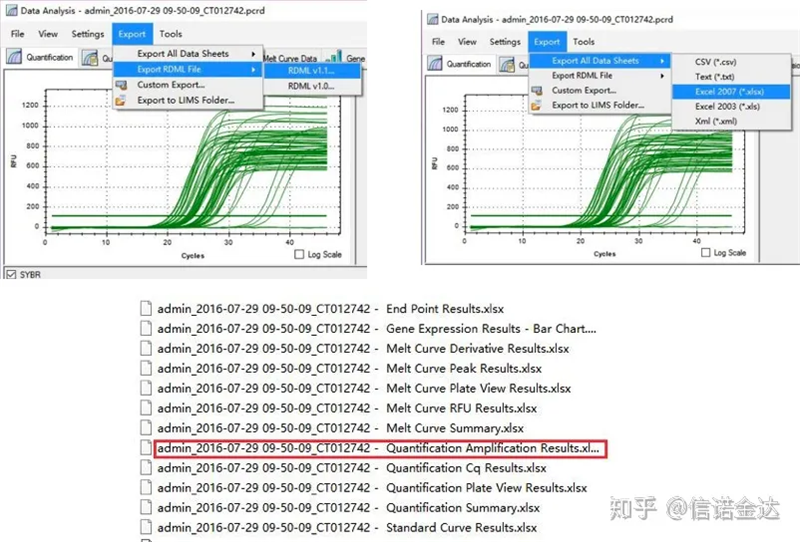
Ryc. 5 Eksport danych qPCR
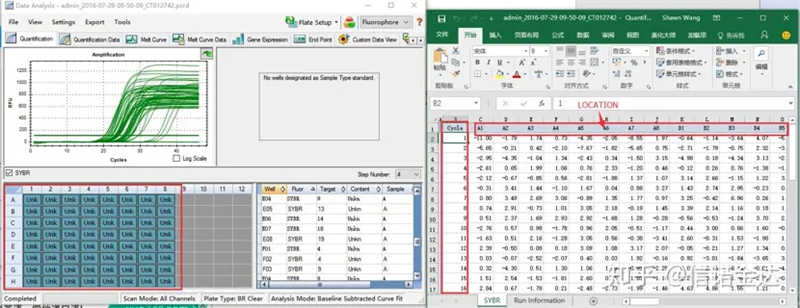
Ryc. 6 wybór próbek kandydujących
Wprowadzanie danych:Otwórz kwalifikację amplifikacji Results.xls, → otwórz LinRegPCR → plik → odczyt z Excela → wybierz parametry, jak pokazano na rysunku 7 → OK → kliknij określ linie bazowe
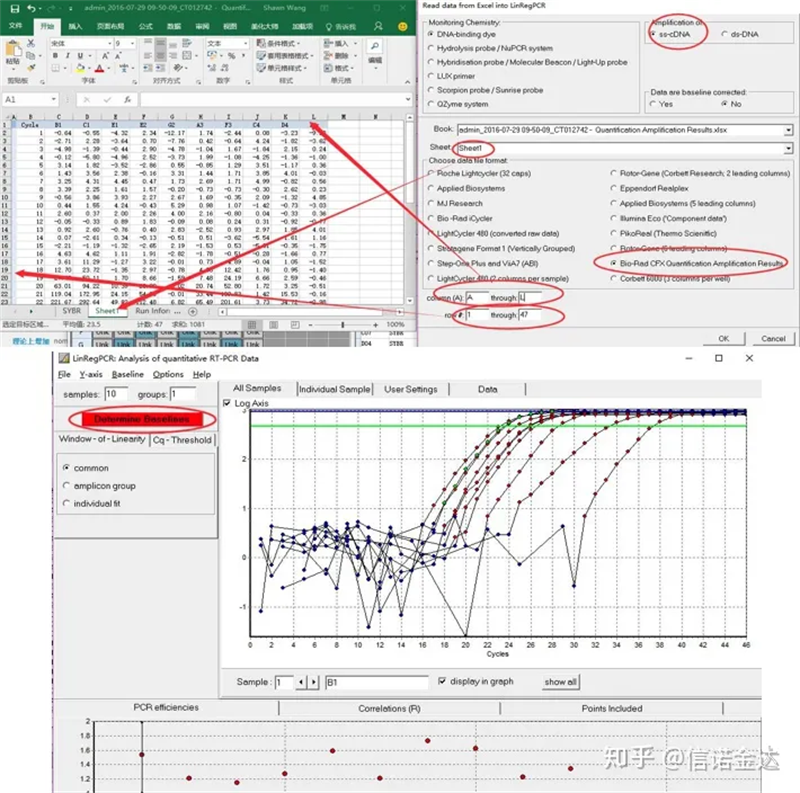
Ryc. 7 etapów wprowadzania danych linRegPCR
Wynik:Jeśli nie ma powtórzeń, grupowanie nie jest wymagane.Jeśli występuje powtórzenie, grupowanie można edytować w grupowaniu próbek, aw identyfikatorze wpisać nazwę genu, a następnie ten sam gen zostanie automatycznie zgrupowany.Na koniec kliknij plik, wyeksportuj program Excel i wyświetl wyniki.Zostaną wyświetlone wydajność amplifikacji i wyniki R2 dla każdego dołka.Po drugie, jeśli podzielisz na grupy, zostanie wyświetlona poprawiona średnia wydajność wzmocnienia.Upewnij się, że skuteczność amplifikacji każdego startera wynosi od 85% do 115%.Jeśli jest za duży lub za mały, oznacza to, że skuteczność amplifikacji startera jest słaba.
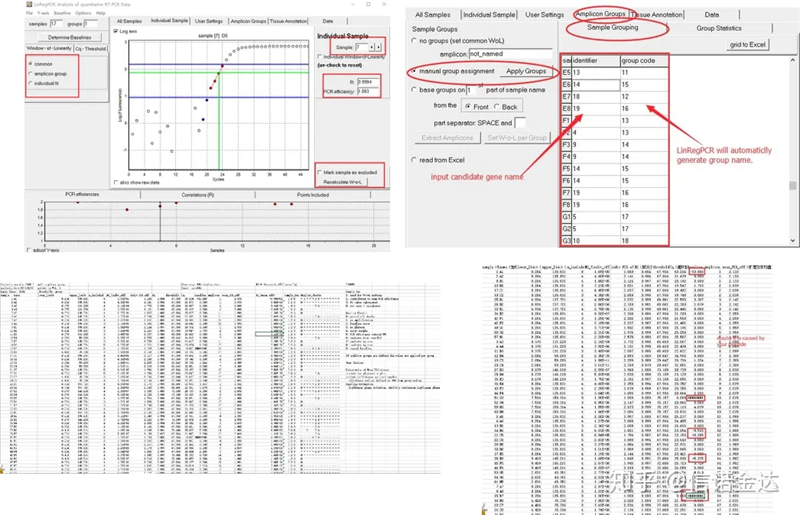
Ryc. 8 Wynik i wyjście danych
Proces eksperymentalny:
Wymagania dotyczące jakości RNA:
Czystość:1.72,0 wskazuje, że mogą występować pozostałości izotiocyjanianu.Czysty kwas nukleinowy A260/A230 powinien wynosić około 2 . Jeśli występuje silna absorpcja przy 230 nm, oznacza to obecność związków organicznych, takich jak jony fenolowe.Ponadto można go wykryć za pomocą elektroforezy w 1,5% żelu agarozowym.Wskaż marker, ponieważ ssRNA nie ma denaturacji, a logarytm masy cząsteczkowej nie ma zależności liniowej, a masa cząsteczkowa nie może być poprawnie wyrażona.Stężenie: Teoretycznieniemniej niż 100 ng/ul, jeśli stężenie jest zbyt niskie, czystość jest na ogół niska, a nie wysoka
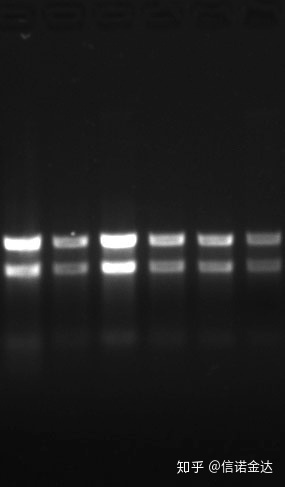
Ryc. 9 żel RNA
Ponadto, jeśli próbka jest cenna, a stężenie RNA jest wysokie, zaleca się podzielenie jej na porcje po ekstrakcji i rozcieńczenie RNA do końcowego stężenia 100-300 ng/ul w celu przeprowadzenia odwrotnej transkrypcji.Wproces odwrotnej transkrypcji, gdy mRNA jest transkrybowany, startery oligo (dt), które mogą specyficznie wiązać się z ogonami poliA, są używane do odwrotnej transkrypcji, podczas gdy lncRNA i circRNA wykorzystują startery losowego heksameru (losowe 6 merów) do odwrotnej transkrypcji całkowitego RNA.Wiele firm wprowadziło teraz specjalne zestawy ogonowe.W przypadku metody łodyga-pętla metoda ogonowania jest wygodniejsza, wydajniejsza i oszczędzająca odczynniki, ale efekt rozróżnienia miRNA z tej samej rodziny nie powinien być tak dobry jak metoda łodygi-pętli.Każdy zestaw do odwrotnej transkrypcji ma wymagania dotyczące stężenia starterów specyficznych dla genu (pętle macierzyste).Wewnętrznym odniesieniem używanym dla miRNA jest U6.W procesie inwersji pnia-pętli, probówkę U6 należy odwrócić oddzielnie, a przedni i tylny starter U6 należy dodać bezpośrednio.Zarówno circRNA, jak i lncRNA mogą wykorzystywać HKG jako wewnętrzne odniesienie.Wwykrywanie cDNA,
jeśli nie ma problemu z RNA, cDNA również powinno być w porządku.Jeśli jednak dąży się do perfekcji eksperymentu, najlepiej jest użyć wewnętrznego genu referencyjnego (gen referencyjny, RG), który może odróżnić gDNA od cds.Ogólnie rzecz biorąc, RG jest genem porządkowym., HKG), jak pokazano na fig. 10;W tym czasie robiłem białko zapasowe z soi i używałem intronów zawierających aktynę7 jako wewnętrznego odniesienia.Wielkość zamplifikowanego fragmentu tego startera w gDNA wynosiła 452 pz, a jeśli cDNA zastosowano jako matrycę, wynosiła 142 pz.Następnie wyniki testu wykazały, że część cDNA była rzeczywiście zanieczyszczona gDNA, a także dowiodły, że nie ma problemu z wynikiem odwrotnej transkrypcji i można go użyć jako matrycy do PCR.Nie ma sensu przeprowadzać elektroforezy w żelu agarozowym bezpośrednio z cDNA i jest to prążek rozproszony, co nie jest przekonujące.
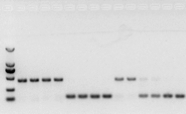
Ryc. 10 Wykrywanie cDNA
Określenie warunków qPCRgeneralnie nie stanowi problemu zgodnie z protokołem zestawu, głównie w kroku wartości tm.Jeśli niektóre startery nie zostały dobrze zaprojektowane podczas projektowania starterów, co skutkuje dużą różnicą między wartością tm a teoretyczną 60°C, zaleca się, aby cDNA Po wymieszaniu próbek przeprowadzić gradientową reakcję PCR ze starterami i starać się unikać ustawiania temperatury bez prążków jako wartości TM.
Analiza danych
Konwencjonalna metoda przetwarzania ilościowego PCR z względną fluorescencją jest zasadniczo zgodna z 2-ΔΔCT.Szablon przetwarzania danych.
Produkty powiązane:
Łatwy PCR w czasie rzeczywistymTM –Taqman
Łatwy PCR w czasie rzeczywistymTM –SYBR ZIELONY I
RT Easy I (Master Premix do syntezy pierwszej nici cDNA)
RT Easy II (Master Premix do syntezy pierwszej nici cDNA do qPCR)
Czas postu: 14 marca 2023 r



