



Jako nowy w laboratorium, nie jest dobrym zadaniem odsiewanie pozytywnych roślin z grupy roślin o niskim współczynniku konwersji.Po pierwsze, DNA musi zostać wyekstrahowane z dużej liczby próbek jedna po drugiej, a następnie obce geny zostaną wykryte metodą PCR.Jednak wyniki są często puste i czasami zawierają kilka elementów, ale nie można określić, czy są to pominięte lub fałszywe wykrycia..Czy zmierzenie się z takim eksperymentalnym procesem i wynikami jest bardzo bezradne?Nie martw się, brat uczy cię, jak łatwo i dokładnie odfiltrowywać transgeniczne pozytywne rośliny.
Krok 1
Startery do wykrywania projektu

Określ gen endogenny i gen egzogenny do wykrycia zgodnie z próbką do przetestowania i wybierz reprezentatywną sekwencję 100-500 pz w genie do zaprojektowania startera.Dobre startery mogą zapewnić dokładność wyników detekcji i skrócić czas detekcji (patrz dodatek do powszechnie stosowanych starterów do detekcji).
Uwaga: Nowo zaprojektowane startery muszą zoptymalizować warunki reakcji i zweryfikować dokładność, precyzję i granicę wykrywalności wykrywania przed wykrywaniem na dużą skalę.
Krok 2
Zaprojektuj protokół eksperymentalny

Kontrola pozytywna: Użyj oczyszczonego DNA zawierającego docelowy fragment jako matrycy, aby określić, czy system i warunki reakcji PCR są normalne.
Kontrola negatywna/pusta: jako matrycy użyć matrycy DNA lub ddH2O, która nie zawiera docelowego fragmentu, aby wykryć, czy w systemie PCR występuje źródło zanieczyszczenia.
Wewnętrzna kontrola referencyjna: użyj kombinacji starter/sonda endogennego genu badanej próbki, aby ocenić, czy matryca może zostać wykryta metodą PCR.
Ogłoszenie:
Dla każdego testu należy ustawić kontrole pozytywne, negatywne/ślepe i kontrole kontroli wewnętrznej, aby ocenić ważność wyników eksperymentu.
Przygotowanie eksperymentu

Przed użyciem należy obserwować, czy roztwór jest równomiernie wymieszany.W przypadku wytrącenia osadu przed użyciem należy go rozpuścić i wymieszać zgodnie z instrukcją.Mieszankę 2×PCR należy przed użyciem pipetować i wielokrotnie mieszać za pomocą mikropipety, aby uniknąć nierównomiernego rozkładu jonów.
Ogłoszenie:
Wyjmij instrukcję i przeczytaj ją uważnie, a przed eksperymentem przygotuj się ściśle według wymagań instrukcji.
Krok 4
Przygotuj system reakcji PCR

Zgodnie z protokołem doświadczalnym zmieszać równomiernie startery, H2O i 2×PCR, odwirować i rozprowadzić je w każdej probówce reakcyjnej.
Ogłoszenie:
W przypadku testów na dużą skalę lub długotrwałych zaleca się stosowanie systemu reakcji PCR zawierającego enzym UNG, który może skutecznie zapobiegać zanieczyszczeniu aerozolem powodowanemu przez produkty PCR.
Krok 5
Dodaj szablon reakcji

Korzystając z technologii Direct PCR, nie ma potrzeby żmudnego procesu oczyszczania kwasu nukleinowego, szablon próbki można przygotować w ciągu 10 minut i można dodać odpowiedni system reakcji PCR.
Ogłoszenie:
Metoda rozszczepiania ma lepszy efekt detekcji, a otrzymany produkt może być wykorzystany do wielu reakcji detekcji.

5.1: Bezpośrednia ekspansja liści
Zgodnie z rozmiarem rysunku w instrukcji, wytnij tkankę liścia o średnicy 2-3mm i umieść ją w systemie reakcji PCR.
Uwaga: Upewnij się, że fragmenty liści są całkowicie zanurzone w roztworze reakcyjnym PCR i nie dodawaj nadmiernej ilości tkanki liściowej.
5.2: Metoda dzielenia liści
Wytnij tkankę liścia o średnicy 5-7 mm i umieść ją w probówce wirówkowej.Jeśli wybierzesz dojrzałe liście, unikaj używania tkanek głównej żyły liścia.Wpipetować 50 ul lizatu Buffer P1 do probówki wirówkowej, aby upewnić się, że lizat może całkowicie zanurzyć tkankę liścia, umieścić go w termocyklerze lub łaźni metalowej i lizować w temperaturze 95°C przez 5-10 minut.

Dodać 50 ul roztworu neutralizującego Buffer P2 i dobrze wymieszać.Otrzymany lizat można wykorzystać jako matrycę i dodać do systemu reakcji PCR.
Uwaga: Ilość matrycy wynosi od 5-10% systemu PCR i nie powinna przekraczać 20% (na przykład w systemie PCR 20 μl dodać 1-2 μl roztworu do lizy, nie więcej niż 4 μl).
Krok 6
reakcja PCR

Po odwirowaniu probówki do reakcji PCR umieszcza się ją w aparacie PCR w celu amplifikacji.
Ogłoszenie:
Reakcja wykorzystuje nieoczyszczoną matrycę do amplifikacji, więc liczba cykli amplifikacji jest o 5-10 cykli większa niż przy użyciu oczyszczonej matrycy DNA.
Krok 7
Detekcja elektroforetyczna i analiza wyników
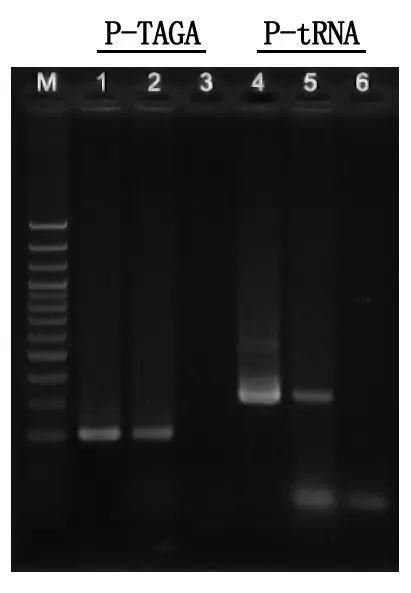
M: drabina DNA 100 pz
1\4: Metoda oczyszczonego DNA
2\5: Bezpośrednia metoda PCR
3\6: Pusta kontrola
kontrola jakości:
Wyniki testów różnych kontroli ustawionych w eksperymencie powinny spełniać następujące warunki.W przeciwnym razie należy przeanalizować przyczynę problemu i po wyeliminowaniu problemu wykonać ponownie badanie.
Tabela 1. Normalne wyniki testów różnych grup kontrolnych
*Gdy plazmid jest używany jako kontrola pozytywna, wynik testu genu endogennego może być ujemny
Ocena wyniku:
A. Wynik testu endogennego genu próbki jest ujemny, co wskazuje, że DNA nadające się do zwykłego wykrywania metodą PCR nie może zostać wyekstrahowane z próbki lub wyekstrahowany DNA zawiera inhibitory reakcji PCR i DNA należy ponownie wyekstrahować.
B. Wynik testu genu endogennego w próbce jest dodatni, a wynik testu genu egzogennego jest ujemny, co wskazuje, że z próbki wyekstrahowano DNA odpowiednie do zwykłego wykrywania PCR i można stwierdzić, że gen XXX nie został wykryty w próbce.
C. Wynik testu genu endogennego w próbce jest pozytywny, a wynik testu genu egzogennego jest pozytywny, co wskazuje, że z próbki wyekstrahowano DNA odpowiednie do zwykłego wykrywania metodą PCR, a DNA próbki zawiera gen XXX.Eksperymenty potwierdzające mogą być dalej przeprowadzane.
Krok 8
Startery do wykrywania projektu

Po eksperymencie użyj 2% roztworu podchlorynu sodu i 70% roztworu etanolu, aby wytrzeć obszar eksperymentalny, aby zapobiec zanieczyszczeniu środowiska.
Czas postu: 08-09-2021



